
Задания
Версия для печати и копирования в MS Word
Задания Д7 № 17459 

i
К какому типу изменчивости относятся ошибки, возникающие при репликации ДНК?
1) мутационная
2) модификационная
3) комбинативная
4) репликационная
Спрятать пояснение
Пояснение.
Мутации – изменения, которые возникают в генетическом аппарате и передаются по наследству. Они бывают спонтанные и индуцированные. Мутации, возникающие без целенаправленного экспериментального вмешательства, называют спонтанными. Как правило, спонтанные мутации можно объяснить случайными ошибками при репликации ДНК.
Раздел кодификатора ФИПИ: 3.6 Закономерности изменчивости. Ненаследственная изменчивость. Норма реакции
Спрятать пояснение
·
Помощь
О проекте · Редакция · Правовая информация · О рекламе
© Гущин Д. Д., 2011—2023
Обновлено: 09.06.2023
Генные мутации присутствуют в каждом человеке. Ученые определили среднее число мутантных генов и скорость возникновения мутаций, которая составляет 100–200 мутаций на поколение.
При слове «мутация» в сознании возникают либо страшноватые образы двухголовых козочек, либо фантастические сверхсущества из фильма «Люди Х». Однако на самом деле в мутациях нет ничего необычного. Без преувеличения можно сказать, что мы все мутанты. Вопрос только в том, какой процент мутировавших генов содержит наша ДНК.
(англ. John Burdon Sanderson Haldane; сокращённо Дж. Б. С. Холдейн, англ. J. B. S. Haldane; 5 ноября 1892, Оксфорд,Оксфордшир, Великобритания — 1 декабря 1964, Бхубанешвар, штат Орисса, Индия) — английский биолог (генетик.
Первая попытка рассчитать темпы мутации генома человека была предпринята в 1935 году одним из отцов современной генетики англичанином Джоном Холдейном. При обследовании мужчины, больного гемофилией, он пришел к выводу, что только в одном из 50000 случаев мутация гена вызывает гемофилию. Это соответствует мутации одного из 25 млн нуклеотидов генома. После Холдейна скорость возникновения мутаций пытались определить, сравнивая ДНК человека и шимпанзе, однако, конечно, точные данные не были получены.
Однако возможности современной генетики позволяют получить точные данные о скорости мутаций — их приводит интернациональная группа из 16 ученых в работе, опубликованной в Current Biology. Они показали, что приблизительные данные, полученные Холдейном 70 лет назад, оказались не так уж далеки от реальности.
стойкое (то есть такое, которое может быть унаследовано потомками данной клетки или организма) изменение генотипа, происходящие под влиянием внешней или внутренней среды. Процесс возникновения мутаций получил название мутагенеза.
Каждый человек является носителем одной мутации в каждых 15–30 млн нуклеотидов.
Для расчета скорости возникновения мутаций авторы работы изучили фрагмент ДНК двух мужчин из китайской деревни, чьи предки несколько сот лет жили в этом же регионе. Общий предок этих мужчин отделен от них 13 поколениями и жил около 200 лет назад. Для чистоты эксперимента ученые исследовали фрагмент мужской Y-хромосомы. Она состоит из 10149085 пар нуклеотидов и передается от отца к сыну в неизменном виде (Y-хромосома у женщин отсутствует). Используя современные методы расшифровки генома, ученые установили, что 10149073 нуклеотидные пары у мужчин не отличимы, то есть всего было локализовано 12 мутаций. Восемь из них при дальнейшем исследовании оказались возникшими уже в клетках взрослого человека в результате их жизнедеятельности, а четыре оказались истинными мутациями, возникшими из-за «ошибки» при передаче генетического материала от отца к сыну.

Приняв эти данные за средние по всему геному и пересчитав их на общее количество генов (полный геном содержит более трех миллиардов нуклеотидов) и 13 поколений, разделяющих мужчин,
ученые рассчитали темпы появления мутаций в человеческом геноме: 100–200 мутаций на поколение.
Большая часть этих мутаций являются безвредным и, в принципе, не заметными для человека, для его организма и здоровья. Однако в редких случаях мутации могут приводит либо к врожденным тяжелым болезням — например, к раку или диабету, либо вносить «усовершенствования» в организм, делая его более стойким.

Интерес к возникновению мутаций и темпам их роста отнюдь не является праздным. Главная их роль — отнюдь не возникновение неизлечимой болезни у какого-либо конкретного человека. Мутации являются необходимым для движения эволюции материалом. Именно они дают генетическое разнообразие, которое позволяет живому миру двигаться вперед. Конечно, на одном-двух поколениях проследить эволюцию невозможно, однако именно мутации дают изменение генома, которое, в случае выгодности его для организма, повышает его стойкость. Если мутация выгодна, то именно носители такого мутантного гена выживают поколение за поколением, в конце концов скрещиваются, и мутация закрепляется уже как системное изменение.
Поэтому изучение скорости и механизма возникновения мутаций может позволить распутать цепь эволюции с конца, как клубок, и прояснить «белые пятна» в истории происхождения видов.
X Международная студенческая научная конференция Студенческий научный форум — 2018

ГЕНОМНЫЕ ХРОМОСОМНЫЕ МУТАЦИИ В ГАМЕТОГЕНЕЗЕ И ЭМБРИОГЕНЕЗЕ ЧЕЛОВЕКА
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке «Файлы работы» в формате PDF
Цель: понять причины возникновений хромосомных мутаций в гаметогенезе и эмбриогенезе человека.
1. Понять, что такое хромосомные мутации.
2. Разобрать причины их возникновений.
3. Выяснить к каким патологиям приводят хромосомные мутации.
1.Что такое геномные хромосомные мутации. 4
2. Геномные мутации. 5
2.1 Полплоидия. 6
2.2 Анэуплоидия. 7
Хромосомные мутации. 8
Список использованной литературы. 14
В современной медицине при изучении болезней человека важное значение имеют наследственные заболевания, связанные с нарушениями целостности генетической информации в геноме человека, вызванными различными факторами.
Геном — наследственный аппарат клетки, содержащий весь объём информации, необходимый для её существования в условиях среды и передачи наследственных признаков последующему поколению.
Геномика – наука, изучающая геном человека и геномы в целом.
Медицинская генетика – одно из направлений геномики человека, система знаний о роли генетических факторов в патологии человека и система методов диагностики, лечения и профилактики наследственной патологии в широком смысле.
Эта наука играет важную роль в профилактической медицине, позволяя посредством различных методов генной терапии предупредить рождение больного ребёнка в семье с наследственной патологией.
Генная терапия предполагает лечение самых разнообразных, а не только наследственных болезней с помощью введения больному генов, играющих ключевую роль в патогенезе соответствующих заболеваний.
Медицинская генетика имеет важное значение в профилактике и лечении любых заболеваний, связанных с какими либо генетическими нарушениями, не только наследственного характера.
1. Что такое геномные хромосомные мутации?
Материальной субстанцией наследственности являются молекулы ДНК и, в частности гены – транскрибируемые фрагменты ДНК, кодирующие белки и разнообразные молекулы РНК (рРНК, тРНК, регуляторные и другие РНК). Изменчивость определяется существованием различных состояний генов или аллелей. При этом нормальная изменчивость связана с присутствием у разных индивидуумов нормальных вариантов гена, а патологическая изменчивость – с наличием множества мутантных аллелей или мутаций. Носители хромосомных аномалий, доминантных мутаций или гомозиготы по рецессивным мутациям называются мутантными особями или мутантами. Мутации называются «легкими» или «тяжелыми», если они ассоциированы с мягким или тяжелым течением заболевания соответственно.
Мутации бывают геномными, хромосомными или генными. В общем случае, геномные и хромосомные мутации приводят к тяжелым патологическим состояниям, часто несовместимым с жизнью.
К геномным мутациям относятся увеличения полного набора хромосом – полиплоидии, или изменения количества хромосом одной пары – анеуплоидии. У человека описано два вида полиплоидий – триплоидии и тетраплоидии – трех- и четырехкратное увеличение числа гаплоидного набора. Подобные аномалии встречаются только у спонтанных абортусов или мертворожденных. [4 c.55]
Полиплоидия (эуплоидия) – геномная мутация, вызванная добавлением целого гаплоидного набора хромосом как в результате ошибок в процессе мейоза, так и при нарушении митоза.
Гаметы и соматические клетки с увеличенным набором хромосом, кратным их гаплоидному числу, называют полиплоидными. Приставки три-, тетра- и т. д. указывают, во сколько раз увеличено число хромосом, т. е. степень плоидности:3n-триплоид, 4n-тетраплоид, 5n-пентаплоид и т. д.
У растений полиплоидия встречается гораздо чаще, чем у животных. Относительная редкость полиплоидии у животных объясняется тем, что увеличение числа хромосом значительно повышает вероятность ошибок при образовании гамет в мейозе.
У человека описаны триплоидные и тетраплоидные организмы. Частота их возникновения низка. Они обнаруживаются среди спонтанно абортированных эмбрионов или плодов и у мертворожденных. Продолжительность жизни новорожденных с такими нарушениями — несколько дней.
Триплоидия может быть обусловлена нарушением мейотического расхождения всего набора хромосом в мейозе женских (отсутствие первого мейотического деления ооцита) или мужских половых клеток. В результате либо яйцеклетка, либо сперматозоид оказываются диплоидными. В качестве механизма триплоидии рассматривают также возможность оплодотворения яйцеклетки двумя сперматозоидами. В том случае, когда триплоидия обусловлена отцовским диплоидным набором хромосом, возникает пузырное перерождение плаценты, так называемый пузырный занос, препятствующий стабильному поступлению питательных веществ от матери к ребенку, а следовательно, его нормальному развитию.
Гораздо больший интерес у учёных-генетиков вызывают заболевания, причиной которых является анэуплоидия.
Анэуплоидия может выражаться в появлении в дочерних клетках добавочной хромосомы (n+1),(2n+1) или в нехватке какой-либо хромосомы (n-1),(2n-1) и т.д. Анэуплоидия может возникнуть, если в анафазе I мейоза гомологичные хромосомы одной или нескольких пар не разойдутся. В этом случае оба члена пары направляются к одному и тому же полюсу клетки, и тогда разделение гомологичных хромосом в анафазе II может привести к образованию гамет, содержащих на одну или несколько хромосом больше или меньше, чем в норме. Это явление известно под названием нерасхождения. Когда гамета с недостающей или лишней хромосомой сливается с нормальной гаплоидной гаметой, образуется зигота с нечетным числом хромосом: вместо каких-либо двух гомологов в такой зиготе их может быть три или только один.
Зигота, в которой число хромосом меньше диплоидного, обычно не развивается, но зиготы с лишними хромосомами иногда способны к развитию. Если это происходит у животных, то из таких зигот в большинстве случаев развиваются особи с резко выраженными аномалиями. У человека наиболее ярким примером нерасхождения хромосом является синдром Дауна, трисомия по 21-й паре хромосом.
Возможно также нерасхождение мужских и женских половых хромосом, которое приводит к анэуплоидии, влияющей на вторичные половые признаки и фертильность, а иногда и на умственные способности.
Хромосомные мутации, в свою очередь, могут быть числовыми (анеуплоидии) или структурными, то есть затрагивать число хромосом или их структуру. Наиболее частыми числовыми аномалиями являются моносомии – отсутствие одной из гомологичных хромосом и трисомии – существование добавочной третьей копии одной из гомологичных хромосом, причем эта добавочная хромосома может быть как материнского, так и отцовского происхождения. Трисомии найдены не для всех хромосом, и наиболее частыми из них являются синдромы Дауна, Эдвардса и Патау – трисомии по 21, 18 и 13 хромосомам соответственно. Иногда количество добавочных хромосом может быть еще больше, эти аномалии называются полисомиями. Моносомии и полисомии описаны, главным образом, для половых хромосом. Другие геномные мутации несовместимы с жизнью и приводят к ранней эмбриональной гибели.[4 c.56]
Синдром Шерешевского Тернера-заболевание ,вызванное моносомией по Х-хромосоме(45 хромосом = 44 аутосомы + ХО).
В период созревания гамет наблюдаются случаи нерасхождения половых хромосом (в I, II или обоих делениях созревания).
Гаметы несут не 22 аутосомы + 1половую хромосому (Х или У), а возникает нарушение парности хромосом. Моносомия Х зависит исключительно от отца.
Р ♀ 44 аутосомы + ХХ → ♂ 44 аутосомы + XY
Гаметы 22 аутосомы + Х 22 аутосомы + ХY
22 аутосомы + Х 22 аутосомы + 0
F144 аутосомы + Х0
Для женщин с синдромом Шерешевского-Тернера характерны маленький рост, короткая шея, воронкообразная грудина, бесплодие вследствие недоразвития яичников, слабое развитие половых признаков. 50% больных умственно отсталы или нормальны. Могут быть пороки развития внутренних органов. Дети с синдромом Шерешевского-Тернера рождаются с частотой 0,7 на 1000 новорожденных девочек.
Диагноз ставят при исследовании полового хроматина и на основании результатов цитогенетического анализа.
Аутосомные моносомии среди живорожденных очень редки. Это мозаичные организмы с нормальными клетками. Моносомия касается аутосом 21 и 22. Полные трисомии описаны по большому числу хромосом: 8, 9, 13, 14, 18, 21, 22 и Х. Число Х-хромосом у человека может доходить до 5 с сохранением жизнеспособности.
Изменение числа хромосом вызвано нарушением распределения их по дочерним клеткам во время 1-го или 2-го мейотического деления в гаметогенезе или при первых дроблениях оплодотворенной яйцеклетки.
— при расхождении во время анафазы редуплицированной хромосомы, в результате чего удвоенная хромосома попадает только в одну дочернюю клетку;
— при нарушении конъюгации гомологичных хромосом, что может нарушить правильность расхождения гомологов по дочерним клеткам;
— при отставании хромосом в анафазе при их расхождении в дочерние клетки, что может привести к утрате хромосомы.
При нарушении в двух и более последовательных делениях возникают тетрасомии и другие полисомии.
К полисомиям относятся синдром Клайнфельтера, синдром Тернера и трисомия по Х-хромосоме.
Женщины с кариотипом ХХХ встречаются с частотой 1-1,4 на 1000 родившихся девочек. Для больных с кариотипом ХХХ характерно наличие недоразвитых яичников, матки, бесплодие. Умственное развитие нормальное или в пределах нижней границы нормы. Около 30% женщин сохраняют способность иметь детей.
С увеличением числа Х-хромосом в кариотипе до 4, 5 и более клинические проявления синдрома увеличиваются. Больные не могут иметь детей, умственно более отсталы. При исследовании полового хроматина в ядрах клеток эпителия слизистой оболочки щеки обнаруживают 2 и более телец Барра. Впервые синдром трисомии по Х-хромосоме описал П.Джекобе в 1959 г.
1 Х-хромосома: нормальный мужчина XY или больная женщина ХО (синдром Шерешевского-Тернера)
2 Х-хромосомы: нормальная женщина ХХ или больной мужчина XXY (синдром Клайнфельтера)
3 Х-хромосомы: больная женщина ХХХ или больной мужчина ХХХY (синдром Клайнфельтера)
4 Х-хромосомы: больная женщина(полисомия Х) или больной мужчина XXXY (синдром Клайнфельтера)
При синдроме Клайнфельтера, описанном им в 1942 г., у мужчин в ядрах клеток эпителия слизистой оболочки полости рта обнаружено тельце Барра. В кариотипе 47 хромосом (44+XXY). Частота больных с синдромом Клайнфельтера колеблется в пределах 2-2,5 на 1000 новорожденных мальчиков.
Для мужчин с синдромом Клайнфельтера характерен высокий рост, длинные конечности, евнухоидизм, нарушенный сперматогенез и бесплодие, гинекомастия, повышенное выделение женских гормонов, склонность к ожирению. Иногда наблюдается антисоциальное поведение и алкоголизм. Степень тяжести симптомов пропорциональна числу добавочных Х-хромосом.
Разновидностью синдрома Клайнфельтера является полисомия по хромосоме Y – синдром XYY (47 хромосом). У мужчин с хромосомным набором XYY рост выше среднего, умственное развитие ниже нормы. Они отличаются агрессивным поведением, наблюдается бесплодие. Среди новорожденных мальчики с данным синдромом рождаются с частотой 1:1000.
Полные трисомии описаны по большому числу аутосом: 8, 9, 13,14, 18, 21, 22.
Трисомия по хромосоме 8 приводит к живорождению, но часто наблюдается мозаицизм. Рождение детей с этим геномным нарушением происходит с частотой 1:50000 новорожденных. При синдроме отмечается неглубокая умственная отсталость и физическое недоразвитие. Типичны скелетные аномалии, удлиненное туловище, нарушения речи.
Трисомия по 9-й паре хромосом заканчивается внутриутробной гибелью носителя лишней хромосомы. Продолжительность жизни немногих рожденных детей с такой трисомией-9 составляла 3,5 месяца. Для них характерны внутриутробное недоразвитие, черепно-лицевые пороки, аномалии скелета, пороки сердца, почек и других органов.
Трисомия по 13-й паре хромосом (синдром Патау) — была описана в 1960 г. – встречается с частотой 1:5000-7000.
Для синдрома характерны пороки, лица, внутренних органов (сердца, почек, половых органов), полидактилия. Глухота наблюдается в 80-85% случаев. Имеет место ранняя смертность (в течение года 90% больных).
Трисомии по 14-й паре хромосом описаны для мертворожденных. У живорожденных этой патологии не выявлено.
Наиболее часто встречается трисомия по 21-й паре хромосом (синдром Дауна). Клиническое описание этого синдрома было сделано в 1866 г. Английским врачом Дауном. Мальчики и девочки заболевают одинаково часто. Частота рождения детей с синдромом Дауна – 1:700-800 новорожденных. В большинстве случаев при трисомии в кариотипе 47 хромосом.
Больные с синдромом Дауна небольшого роста, слабоумны, имеют физические пороки. Для них характерны небольшая голова со скошенным затылком, косые глазные щели, эпикант, короткий нос с широкой переносицей, маленькие деформированные уши, полуоткрытый рот с высунутым языком и выступающей челюстью, походка с неловкими движениями, косноязычие. Они имеют пороки сердца, желудочно-кишечного тракта, почек. У больных часто возникают инфекционные и злокачественные заболевания, что обусловлено дефектами иммунной системы. Особенности дерматоглифики связаны с глубокой поперечной бороздой (обезьянья складка) и единственной сгибательной складкой на мизинцах. Благодаря улучшению условий жизни и медицинской помощи, больные с синдромом Дауна доживают до 30 лет и более. Некоторые больные могут заниматься посильной трудовой деятельностью.
Трисомия по 22-й паре, как правило, вызывает летальный эффект и гибель плода во внутриутробном периоде.
Такие генетические нарушения как эуплоидия и анэуплоидия относятся к числу геномных мутаций, т.е. мутаций, связанных с изменением первоначального числа хромосом в клетке.
Необходимо научиться предотвращать данные заболевания во избежание возникновения тяжелых обширных патологий в еще не сформировавшемся человеческом организме. Высокая смертность среди новорожденных с генетическими отклонениями, описанными в данной работе, связана с нарушением развития плода на ранних этапах жизни.
Очень часто большую роль в возникновении мутаций играет человеческий фактор, т.к. загрязнение окружающей среды, нездоровый образ жизни, вредные привычки, такие как курение и алкоголизм пагубно сказываются не только на здоровье человека, но и на здоровье его потомства.
Список использованной литературы
Гинтер Е.К. Медицинская генетика. М.: Медицина, 2008.
Грин Н., Стаут У., Тейлор Д. Биология т. 1-3. М.: Мир, 2009.
В.А. Шевченко, Н.А. Топорнина, Н.С. Стволинская Генетика человека. М.: Владос, 2007.
Горбунова В. Н. Медицинская генетика, 2009
Л.А. Рязанова, Учебное пособие, Практические занятия по основам генетики, 2014
Происхождение мутаций: геномные, хромосомные, генные мутации
Происхождение мутаций: геномные, хромосомные, генные мутации
Геномные мутации. Нерасхождение пары хромосом в ходе мейоза вызывает геномные мутации, например трисомию 21 (синдром Дауна). Геномные мутации приводят к хромосомным анеуплоидиям и бывают наиболее частыми мутациями у человека, с частотой 1 случай нерасхождения на 25-50 мейотических делений клетки.
Это минимальная оценка, поскольку последствия большинства таких мутаций настолько серьезны, что анеуплоидные эмбрионы спонтанно прерываются вскоре после зачатия. Геномные мутации также часто выявляют в клетках опухолей.
Хромосомные мутации
Хромосомные мутации, происходящие с частотой приблизительно одна перестройка на 1700 клеточных делений, случаются значительно реже геномных мутаций. Хотя частоты геномных и хромосомных мутаций могут казаться высокими, эти мутации редко передаются от одного поколения следующему, поскольку они обычно несовместимы с жизнью или нормальной репродукцией. Хромосомные мутации также часто обнаруживают в клетках опухолей.
Генные мутации
Генные мутации, включая замены пар оснований, вставки и делеции, возникают по одному из двух основных механизмов: ошибок в нормальном процессе репликации ДНК или вследствие нарушения репарации ДНК после повреждения. Некоторые мутации происходят спонтанно, другие вызываются физическими или химическими агентами, названными мутагенами, поскольку они существенно повышают частоту мутаций.

Ошибки репликации ДНК. Большинство ошибок репликации быстро удаляются из ДНК и корректируются комплексом ферментов репарации ДНК, сначала опознающим, какая из нитей вновь синтезированной двойной спирали содержит неправильное основание, а затем заменяющим его соответствующим комплементарным основанием.
Репарация ДНК должна быть в высшей степени точным процессом; в противном случае число мутаций в организме было бы недопустимым, и наш вид перестал бы существовать. Фермент ДНК-полимераза точно дублирует двойную спираль благодаря строгому правилу комбинации пар оснований (А с Т, С с G) и молекулярной корректировке.
Всего один неправильный нуклеотид попадает в одну из растущих дочерних нитей на 10 миллионов пар оснований (и это при перемещении вдоль хромосомы человека со скоростью около 50 пар оснований в секунду!). Дополнительная проверка ошибок затем корректирует более 99,9% ошибок репликации ДНК. Таким образом, общий показатель мутаций в результате ошибок репликации имеет в высшей степени низкий уровень 10-10 на пару оснований за одно деление клетки.
Поскольку человеческий диплоидный геном содержит приблизительно 6х109 пары оснований ДНК, репликация ошибок приводит менее чем к одной новой мутации пар оснований на деление клетки.
Репарация повреждений ДНК
Считают, что кроме ошибок репликации, от 10 000 до 1 000 000 нуклеотидов на клетку в день повреждаются спонтанными химическими процессами, такими как, например, деметилирование или деаминирование, реакциями с химическими мутагенами (природными или иными) среды и влиянием ультрафиолетового или ионизирующего излучения.
Некоторые, но не все из этих дефектов могут быть исправлены. Даже если повреждение обнаружено и удалено, система репарации может неточно прочитать комплементарную нить и, как следствие, создать мутацию, вводя неправильные основания. Таким образом, в отличие от изменений ДНК, связанных с репликацией, которые обычно корректируются репарационным механизмом, изменения нуклеотидов, возникающие при репарации поврежденной ДНК, часто приводят к стойким мутациям.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Мутации
Автор статьи — Л.В. Окольнова.
Сразу на ум приходят Люди Х… или Человек — Паук …
Но это в кино, в биологии тоже так, но немного более научно, менее фантастично и более обыденно.
Мута́ция (в переводе — изменение) — устойчивое, передающееся по наследству изменение ДНК, происходящее под влиянием внешних или внутренних изменений.
Мутагенез — процесс появления мутаций.
Обыденность в том, что эти изменения (мутации) происходят в природе и у человека постоянно, почти каждодневно.
В первую очередь, мутации подразделяются на соматические — возникают в клетках тела, и генеративные — появляются только в гаметах.
Соматические мутации
Генеративные мутации
Передаются по наследству.
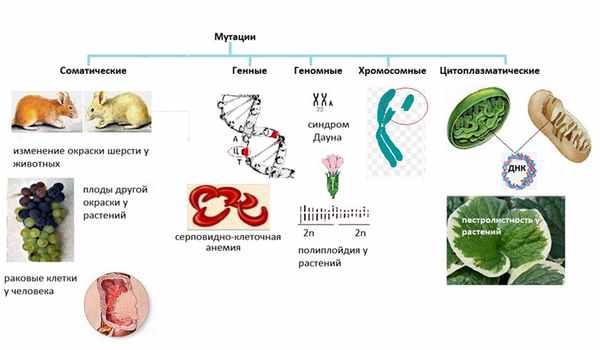
Разберем сначала виды генеративных мутаций.
Генные мутации
Что такое ген? Это участок ДНК (т.е. несколько нуклеотидов), соответственно, это и участок РНК, и участок белка, и какой-либо признак организма.
Т.е. генная мутация — это выпадение, замена, вставка, удвоение, изменение последовательности участков ДНК.
Вообще, это не всегда ведет к болезни. Например, при удвоении ДНК случаются такие “ошибки”. Но они возникают редко, это очень малый процент от всего количества, поэтому они незначительны, что практически не влияют на организм.
Бывают и серьезные мутагенезы:
— серповидно-клеточная анемия у человека;
— фенилкетонурия — нарушение обмена веществ, вызывающее довольно серьезные нарушения умственного развития
— гемофилия
— гигантизм у растений
Геномные мутации
Вот классическое определение термина “геном”:
— совокупность наследственного материала, заключенного в клетке организма;
— геном человека и геномы всех остальных клеточных форм жизни, построены из ДНК;
— совокупность генетического материала гаплоидного набора хромосом данного вида в парах нуклеотидов ДНК на гаплоидный геном.
Для понимания сути мы очень сильно упростим, получится такое определение:
Геном — это количество хромосом
Геномные мутации — изменение числа хромосом организма. В основном, их причина — нестандартное расхождение хромосом в процессе деления.
— синдром Дауна — в норме у человека 46 хромосом (23 пары), однако при этой мутации образуются 47 хромосом
рис. синдром Дауна
— полиплойдия у растений (для растений это вообще норма — большинство культурный растений — полиплойдные мутанты)
Хромосомные мутации — деформации самих хромосом.
Примеры (некоторые перестройки такого рода есть у большинства людей и вообще никак не отражаются ни внешне, ни на здоровье, но есть и неприятные мутации):
— синдром кошачьего крика у ребенка
— задержка в развитии
и т.д.
Цитоплазматические мутации — мутации в ДНК митохондрий и хлоропластов.
Есть 2 органеллы со своими собственными ДНК (кольцевыми, в то время как в ядре — двойная спираль) — митохондрия и растительные пластиды.
Соответственно, есть мутации, вызванные изменениями именно в этих структурах.
Есть интересная особенность — этот вид мутации передается только женским полом, т.к. при образовании зиготы остаются только материнские митохондрии, а “мужские” отваливаются с хвостом при оплодотворении.
Примеры:
— у человека — определенная форма сахарного диабета, туннельное зрение;
— у растений — пестролистность.
Соматические мутации.
Это все описанные выше виды, но возникают они в клетках тела ( в соматических клетках).
Мутантных клеток обычно намного меньше, чем нормальных, и они подавляются здоровыми клетками. (Если не подавляются, то организм перерождаться или болеть).
Примеры:
— у дрозофилы глаз красный, но может иметь белые фасеты
— у растения это может быть целый побег, отличающийся от других (И.В. Мичурин таким образом выводил новые сорта яблок).
— раковые клетки у человека
Примеры вопросов ЕГЭ:
Синдром Дауна является результатом мутации
Генные мутации связаны с изменением
А) числа хромосом в клетках;
Б) структуры хромосом;
B) последовательности генов в аутосоме;
Г) нуклеогидов на участке ДНК.
Мутации, связанные с обменом участками негомологичных хромосом, относят к
Животное, в потомстве которого может появиться признак, обусловленный соматической мутацией
Виды мутаций. Примеры. Причины.

Мутации делятся на несколько основных видов
В биологии все виды мутаций являются одним из эволюционных факторов, так как они могут имеют шанс быть унаследованными потомками мутировавшего организма, микроорганизма или даже клетки, передав эти изменения дальше, что в конечном счёте приведёт к изменению генома. Мутационные процессы, таким образом, оказывают влияние на ход эволюции, причём сами мутации можно разделить на три группы – полезные, нейтральные и вредные. Как показывает изучение закономерностей мутационных процессов, полезных среди них меньше всего, но влияние модификационной изменчивости и этих самых процессов на естественный отбор очень велико.
Геномные мутации
Говоря простым языком, это изменение числа хромосом организма. В основном, их причина – нестандартное расхождение хромосом в процессе деления.
- Пример геномной мутации у человека: синдром Дауна, который вызывается наличием “лишней” хромосомы. У человека с этим синдромом хромосом не 46, а 47.
- Пример геномной мутации у растений: полиплойдия. Но в ней нет ничего страшного – например, большинство культурных растений имеют эту мутацию.
Генные мутации
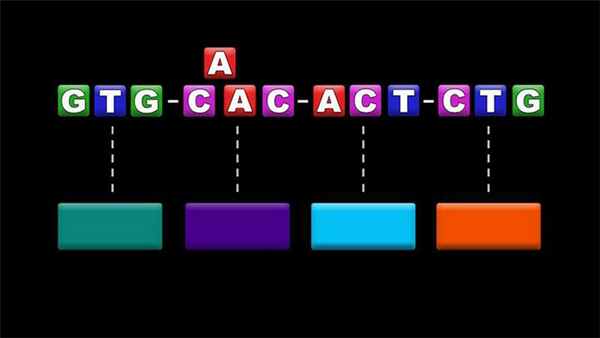
Изменение нуклеотидной последовательности приводит к генной мутации
Это изменение нуклеотидной последовательности в одной молекуле ДНК. При этом в том случае, когда под действием мутации изменяется лишь один нуклеотид, мутация называется точечной. При точечной мутации одно азотистое основание в ДНК или РНК заменяется другим. Если же один участок ДНК заменяется участком другой длины и другого нуклеотидного состава, то такая генная мутация уже называется не точечной, а сложной. Строго говоря, к генной мутации может приводить не только замена одного нуклеотида другим, но и просто вставка другого нуклеотида (без выпадения прежнего) или, наоборот, выпадение нуклеотида без его замены на другой.
Пример генной мутации у растений
- Гигантизм у растений. Возникает он именно из-за генной мутации.
Пример генной мутации у животных и человека
- Фенилкетонурия. Это встречающееся у людей наследственное заболевание, кстати, одно из немногих, поддающихся успешному лечению. Оно связано с нарушением метаболизма аминокислот, главным образом фенилаланина.

Какое влияние оказала научная революция на европейское чудо

30 интересных фактов о Каспийском море
Хромосомные мутации
В отличие от геномных мутаций, они возникают не из-за “неправильного” числа хромосом, а из-за их повреждения. При этом у многих людей можно найти признаки хромосомных мутаций, которые при этом никак не влияют ни на их здоровье, ни на внешность, ни на умственное развитие. Но в некоторых случаях из-за этого мутационного процесса могут возникать хромосомные болезни, которые приводят к наследственным заболеваниям. При этом хромосомные мутации делятся на две категории:
- Внутрихромосомные. Это преобразование генетического материала в пределах одной хромосомы.
- Межхромосомные. Это перестройки, в результате которых две негомологичные хромосомы обмениваются своими участками. Негомологичные хромосомы содержат разные гены и не встречаются в процессе мейоза.
Мутации в соматических клетках
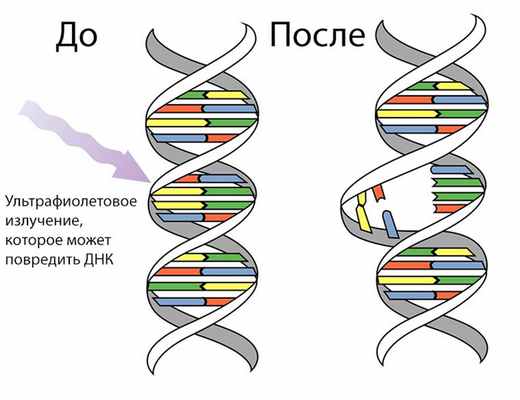
Ультрафиолетовое излучение – одна из причин соматических мутаций
Как мы знаем из биологии, к соматическим клеткам относятся все клетки тела, не имеющие прямого отношения к процессу полового размножения. Соматические мутации возникают в клетках тела и обусловливают мозаичность организма, то есть образование в нём отдельных участков тела, тканей или клеток с отличным от остальных набором хромосом или генов. При этом у них могут возникать мутации всех типов, что и у половых клеток. Одной из их причин, например, могут послужить различные виды излучения. Соматические мутации возникают в соматических клетках, проявляются у данного организма и не передаются потомству при половом размножении. Мутации, возникающие в соматических клетках, наследуются дочерними клетками, которые образуются в процессе митотических делений.
- Пример соматической мутации: окраска шерсти у валлийских овец, у которых присутствует чёрное пятно под хвостом, отличающееся по цвету от остальной части шерстного покрова. Эта мутация могла проявиться либо как доминантная, либо как рецессивная при потере части или всей гомологичной хромосомы.

Образ и характеристика Печорина в «Герое нашего времени»

25 интересных фактов об Эльбрусе
Генеративные мутации
Это мутации, возникающие в клетках полового зачатка и в половых клетках. Собственно, только это и отличает их от соматических мутаций, так как эволюционная ценность генеративных и соматических мутаций различна, она определяется типом размножения организма. При этом генеративные мутации по своей природе ничем не отличаются от соматических.
- Пример генеративной мутации: синдром Дауна у человека. Значительное количество эмбрионов в таких случаях не выживает вовсе, но те из них, что успешно появляются на свет и остаются в живых в первые годы, имеют все шансы дожить до преклонных лет.
Результаты мутаций
Они могут быть самыми разными, как положительными, так и нейтральными или отрицательными. При этом повлиять на дальнейший эволюционный процесс имеют больше шансов именно положительные мутации, так как они предоставляют живым организмам определённые конкурентные преимущества. Отрицательные мутации, которые ослабляют организм тем или иным образом, напротив, снижают шансы организма-мутанта на выживание.
Свойства мутаций

Мутации могут обладать как положительными свойствами, так и негативными
- Они всегда возникают внезапно.
- Они могут передаваться по наследству.
- Из-за отсутствия у них направленного характера их появление невозможно предсказать.
- Они могут быть как полезными, так и вредными или нейтральными.
- Сходные мутации могут возникать многократно.
Причины мутаций

Загрязнение окружающей среды часто приводит к мутациям
Читайте также:
- Синдром Эйзенменгера
- Жгутиконосцы. Трихомоноз. Возбудитель трихомоноза.
- Тромбоз мезентериальных сосудов. Клиника тромбоза мезентериальных сосудов
- Патанатомия несахарного мочеизнурения. Ядра гипоталамуса влияющие на гипофиз
- Лейкодистрофии. Метахроматическая лейкодистрофия
![]()
![]()
FAQ

Алексей Кондрашов
Сохранить в закладки
26611
26
Сохранить в закладки
6 фактов об ошибках при репликации и репарации ДНК, наследственных болезнях и слабовредных мутациях
19.06.2014
Над материалом работали
Алексей Кондрашов
кандидат биологических наук, профессор кафедры экологии и эволюционной биологии Мичиганского университета (США)

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
ПРОМО Поддержать ПостНауку — значит поддержать просвещение

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
tv Российский биотех: угрозы, перспективы, решения / Ярослав Ашихмин в Рубке ПостНауки

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
FAQ Способны ли человекообразные обезьяны к коллективному убийству?

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
Видео
24819
Фармакокинетика

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
talks Прямая речь: Станислав Дробышевский

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
лекции Животные считают иначе
Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
FAQ Как возникают раковые клетки?

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
Видео
128348
Гейдельбергский человек

Добавить в закладки
Вы сможете увидеть эту публикацию в личном кабинете
Журнал Знакомьтесь: самая главная молекула

A red tulip exhibiting a partially yellow petal due to a mutation in its genes

Mutation with double bloom in the Langheck Nature Reserve near Nittel, Germany
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA.[1] Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA (such as pyrimidine dimers caused by exposure to ultraviolet radiation), which then may undergo error-prone repair (especially microhomology-mediated end joining),[2] cause an error during other forms of repair,[3][4] or cause an error during replication (translesion synthesis). Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.[5][6][7]
Mutations may or may not produce detectable changes in the observable characteristics (phenotype) of an organism. Mutations play a part in both normal and abnormal biological processes including: evolution, cancer, and the development of the immune system, including junctional diversity. Mutation is the ultimate source of all genetic variation, providing the raw material on which evolutionary forces such as natural selection can act.
Mutation can result in many different types of change in sequences. Mutations in genes can have no effect, alter the product of a gene, or prevent the gene from functioning properly or completely. Mutations can also occur in non-genic regions. A 2007 study on genetic variations between different species of Drosophila suggested that, if a mutation changes a protein produced by a gene, the result is likely to be harmful, with an estimated 70% of amino acid polymorphisms that have damaging effects, and the remainder being either neutral or marginally beneficial.[8] Due to the damaging effects that mutations can have on genes, organisms have mechanisms such as DNA repair to prevent or correct mutations by reverting the mutated sequence back to its original state.[5]
Overview[edit]
Mutations can involve the duplication of large sections of DNA, usually through genetic recombination.[9] These duplications are a major source of raw material for evolving new genes, with tens to hundreds of genes duplicated in animal genomes every million years.[10] Most genes belong to larger gene families of shared ancestry, detectable by their sequence homology.[11] Novel genes are produced by several methods, commonly through the duplication and mutation of an ancestral gene, or by recombining parts of different genes to form new combinations with new functions.[12][13]
Here, protein domains act as modules, each with a particular and independent function, that can be mixed together to produce genes encoding new proteins with novel properties.[14] For example, the human eye uses four genes to make structures that sense light: three for cone cell or color vision and one for rod cell or night vision; all four arose from a single ancestral gene.[15] Another advantage of duplicating a gene (or even an entire genome) is that this increases engineering redundancy; this allows one gene in the pair to acquire a new function while the other copy performs the original function.[16][17] Other types of mutation occasionally create new genes from previously noncoding DNA.[18][19]
Changes in chromosome number may involve even larger mutations, where segments of the DNA within chromosomes break and then rearrange. For example, in the Homininae, two chromosomes fused to produce human chromosome 2; this fusion did not occur in the lineage of the other apes, and they retain these separate chromosomes.[20] In evolution, the most important role of such chromosomal rearrangements may be to accelerate the divergence of a population into new species by making populations less likely to interbreed, thereby preserving genetic differences between these populations.[21]
Sequences of DNA that can move about the genome, such as transposons, make up a major fraction of the genetic material of plants and animals, and may have been important in the evolution of genomes.[22] For example, more than a million copies of the Alu sequence are present in the human genome, and these sequences have now been recruited to perform functions such as regulating gene expression.[23] Another effect of these mobile DNA sequences is that when they move within a genome, they can mutate or delete existing genes and thereby produce genetic diversity.[6]
Nonlethal mutations accumulate within the gene pool and increase the amount of genetic variation.[24] The abundance of some genetic changes within the gene pool can be reduced by natural selection, while other «more favorable» mutations may accumulate and result in adaptive changes.

For example, a butterfly may produce offspring with new mutations. The majority of these mutations will have no effect; but one might change the color of one of the butterfly’s offspring, making it harder (or easier) for predators to see. If this color change is advantageous, the chances of this butterfly’s surviving and producing its own offspring are a little better, and over time the number of butterflies with this mutation may form a larger percentage of the population.
Neutral mutations are defined as mutations whose effects do not influence the fitness of an individual. These can increase in frequency over time due to genetic drift. It is believed that the overwhelming majority of mutations have no significant effect on an organism’s fitness.[25][26] Also, DNA repair mechanisms are able to mend most changes before they become permanent mutations, and many organisms have mechanisms for eliminating otherwise-permanently mutated somatic cells.
Beneficial mutations can improve reproductive success.[27][28]
Causes[edit]
Four classes of mutations are (1) spontaneous mutations (molecular decay), (2) mutations due to error-prone replication bypass of naturally occurring DNA damage (also called error-prone translesion synthesis), (3) errors introduced during DNA repair, and (4) induced mutations caused by mutagens. Scientists may also deliberately introduce mutant sequences through DNA manipulation for the sake of scientific experimentation.
One 2017 study claimed that 66% of cancer-causing mutations are random, 29% are due to the environment (the studied population spanned 69 countries), and 5% are inherited.[29]
Humans on average pass 60 new mutations to their children but fathers pass more mutations depending on their age with every year adding two new mutations to a child.[30]
Spontaneous mutation[edit]
Spontaneous mutations occur with non-zero probability even given a healthy, uncontaminated cell. Naturally occurring oxidative DNA damage is estimated to occur 10,000 times per cell per day in humans and 100,000 times per cell per day in rats.[31] Spontaneous mutations can be characterized by the specific change:[32]
- Tautomerism – A base is changed by the repositioning of a hydrogen atom, altering the hydrogen bonding pattern of that base, resulting in incorrect base pairing during replication.[33] Theoretical results suggest that proton tunneling is an important factor in the spontaneous creation of GC tautomers.[34]
- Depurination – Loss of a purine base (A or G) to form an apurinic site (AP site).
- Deamination – Hydrolysis changes a normal base to an atypical base containing a keto group in place of the original amine group. Examples include C → U and A → HX (hypoxanthine), which can be corrected by DNA repair mechanisms; and 5MeC (5-methylcytosine) → T, which is less likely to be detected as a mutation because thymine is a normal DNA base.
- Slipped strand mispairing – Denaturation of the new strand from the template during replication, followed by renaturation in a different spot («slipping»). This can lead to insertions or deletions.
Error-prone replication bypass[edit]
There is increasing evidence that the majority of spontaneously arising mutations are due to error-prone replication (translesion synthesis) past DNA damage in the template strand. In mice, the majority of mutations are caused by translesion synthesis.[35] Likewise, in yeast, Kunz et al.[36] found that more than 60% of the spontaneous single base pair substitutions and deletions were caused by translesion synthesis.
Errors introduced during DNA repair[edit]
Although naturally occurring double-strand breaks occur at a relatively low frequency in DNA, their repair often causes mutation. Non-homologous end joining (NHEJ) is a major pathway for repairing double-strand breaks. NHEJ involves removal of a few nucleotides to allow somewhat inaccurate alignment of the two ends for rejoining followed by addition of nucleotides to fill in gaps. As a consequence, NHEJ often introduces mutations.[37]

Induced mutation[edit]
Induced mutations are alterations in the gene after it has come in contact with mutagens and environmental causes.
Induced mutations on the molecular level can be caused by:
- Chemicals
- Hydroxylamine
- Base analogs (e.g., Bromodeoxyuridine (BrdU))
- Alkylating agents (e.g., N-ethyl-N-nitrosourea (ENU). These agents can mutate both replicating and non-replicating DNA. In contrast, a base analog can mutate the DNA only when the analog is incorporated in replicating the DNA. Each of these classes of chemical mutagens has certain effects that then lead to transitions, transversions, or deletions.
- Agents that form DNA adducts (e.g., ochratoxin A)[39]
- DNA intercalating agents (e.g., ethidium bromide)
- DNA crosslinkers
- Oxidative damage
- Nitrous acid converts amine groups on A and C to diazo groups, altering their hydrogen bonding patterns, which leads to incorrect base pairing during replication.
- Radiation
- Ultraviolet light (UV) (including non-ionizing radiation). Two nucleotide bases in DNA—cytosine and thymine—are most vulnerable to radiation that can change their properties. UV light can induce adjacent pyrimidine bases in a DNA strand to become covalently joined as a pyrimidine dimer. UV radiation, in particular longer-wave UVA, can also cause oxidative damage to DNA.[40]
- Ionizing radiation. Exposure to ionizing radiation, such as gamma radiation, can result in mutation, possibly resulting in cancer or death.
Whereas in former times mutations were assumed to occur by chance, or induced by mutagens, molecular mechanisms of mutation have been discovered in bacteria and across the tree of life. As S. Rosenberg states, «These mechanisms reveal a picture of highly regulated mutagenesis, up-regulated temporally by stress responses and activated when cells/organisms are maladapted to their environments—when stressed—potentially accelerating adaptation.»[41] Since they are self-induced mutagenic mechanisms that increase the adaptation rate of organisms, they have some times been named as adaptive mutagenesis mechanisms, and include the SOS response in bacteria,[42] ectopic intrachromosomal recombination[43] and other chromosomal events such as duplications.[41]
Classification of types[edit]
By effect on structure[edit]
![]()
Five types of chromosomal mutations
![]()
Types of small-scale mutations
The sequence of a gene can be altered in a number of ways.[44] Gene mutations have varying effects on health depending on where they occur and whether they alter the function of essential proteins.
Mutations in the structure of genes can be classified into several types.
Large-scale mutations[edit]
Large-scale mutations in chromosomal structure include:
- Amplifications (or gene duplications) or repetition of a chromosomal segment or presence of extra piece of a chromosome broken piece of a chromosome may become attached to a homologous or non-homologous chromosome so that some of the genes are present in more than two doses leading to multiple copies of all chromosomal regions, increasing the dosage of the genes located within them.
- Polyploidy, duplication of entire sets of chromosomes, potentially resulting in a separate breeding population and speciation.
- Deletions of large chromosomal regions, leading to loss of the genes within those regions.
- Mutations whose effect is to juxtapose previously separate pieces of DNA, potentially bringing together separate genes to form functionally distinct fusion genes (e.g., bcr-abl).
- Large scale changes to the structure of chromosomes called chromosomal rearrangement that can lead to a decrease of fitness but also to speciation in isolated, inbred populations. These include:
- Chromosomal translocations: interchange of genetic parts from nonhomologous chromosomes.
- Chromosomal inversions: reversing the orientation of a chromosomal segment.
- Non-homologous chromosomal crossover.
- Interstitial deletions: an intra-chromosomal deletion that removes a segment of DNA from a single chromosome, thereby apposing previously distant genes. For example, cells isolated from a human astrocytoma, a type of brain tumor, were found to have a chromosomal deletion removing sequences between the Fused in Glioblastoma (FIG) gene and the receptor tyrosine kinase (ROS), producing a fusion protein (FIG-ROS). The abnormal FIG-ROS fusion protein has constitutively active kinase activity that causes oncogenic transformation (a transformation from normal cells to cancer cells).
- Loss of heterozygosity: loss of one allele, either by a deletion or a genetic recombination event, in an organism that previously had two different alleles.
Small-scale mutations[edit]
Small-scale mutations affect a gene in one or a few nucleotides. (If only a single nucleotide is affected, they are called point mutations.) Small-scale mutations include:
- Insertions add one or more extra nucleotides into the DNA. They are usually caused by transposable elements, or errors during replication of repeating elements. Insertions in the coding region of a gene may alter splicing of the mRNA (splice site mutation), or cause a shift in the reading frame (frameshift), both of which can significantly alter the gene product. Insertions can be reversed by excision of the transposable element.
- Deletions remove one or more nucleotides from the DNA. Like insertions, these mutations can alter the reading frame of the gene. In general, they are irreversible: Though exactly the same sequence might, in theory, be restored by an insertion, transposable elements able to revert a very short deletion (say 1–2 bases) in any location either are highly unlikely to exist or do not exist at all.
- Substitution mutations, often caused by chemicals or malfunction of DNA replication, exchange a single nucleotide for another.[45] These changes are classified as transitions or transversions.[46] Most common is the transition that exchanges a purine for a purine (A ↔ G) or a pyrimidine for a pyrimidine, (C ↔ T). A transition can be caused by nitrous acid, base mispairing, or mutagenic base analogs such as BrdU. Less common is a transversion, which exchanges a purine for a pyrimidine or a pyrimidine for a purine (C/T ↔ A/G). An example of a transversion is the conversion of adenine (A) into a cytosine (C). Point mutations are modifications of single base pairs of DNA or other small base pairs within a gene. A point mutation can be reversed by another point mutation, in which the nucleotide is changed back to its original state (true reversion) or by second-site reversion (a complementary mutation elsewhere that results in regained gene functionality). As discussed below, point mutations that occur within the protein coding region of a gene may be classified as synonymous or nonsynonymous substitutions, the latter of which in turn can be divided into missense or nonsense mutations.
By impact on protein sequence[edit]

![]()
Point mutations classified by impact on protein
![]()
The effect of a mutation on protein sequence depends in part on where in the genome it occurs, especially whether it is in a coding or non-coding region. Mutations in the non-coding regulatory sequences of a gene, such as promoters, enhancers, and silencers, can alter levels of gene expression, but are less likely to alter the protein sequence. Mutations within introns and in regions with no known biological function (e.g. pseudogenes, retrotransposons) are generally neutral, having no effect on phenotype – though intron mutations could alter the protein product if they affect mRNA splicing.
Mutations that occur in coding regions of the genome are more likely to alter the protein product, and can be categorized by their effect on amino acid sequence:
- A frameshift mutation is caused by insertion or deletion of a number of nucleotides that is not evenly divisible by three from a DNA sequence. Due to the triplet nature of gene expression by codons, the insertion or deletion can disrupt the reading frame, or the grouping of the codons, resulting in a completely different translation from the original.[48] The earlier in the sequence the deletion or insertion occurs, the more altered the protein produced is. (For example, the code CCU GAC UAC CUA codes for the amino acids proline, aspartic acid, tyrosine, and leucine. If the U in CCU was deleted, the resulting sequence would be CCG ACU ACC UAx, which would instead code for proline, threonine, threonine, and part of another amino acid or perhaps a stop codon (where the x stands for the following nucleotide).) By contrast, any insertion or deletion that is evenly divisible by three is termed an in-frame mutation.
- A point substitution mutation results in a change in a single nucleotide and can be either synonymous or nonsynonymous.
- A synonymous substitution replaces a codon with another codon that codes for the same amino acid, so that the produced amino acid sequence is not modified. Synonymous mutations occur due to the degenerate nature of the genetic code. If this mutation does not result in any phenotypic effects, then it is called silent, but not all synonymous substitutions are silent. (There can also be silent mutations in nucleotides outside of the coding regions, such as the introns, because the exact nucleotide sequence is not as crucial as it is in the coding regions, but these are not considered synonymous substitutions.)
- A nonsynonymous substitution replaces a codon with another codon that codes for a different amino acid, so that the produced amino acid sequence is modified. Nonsynonymous substitutions can be classified as nonsense or missense mutations:
- A missense mutation changes a nucleotide to cause substitution of a different amino acid. This in turn can render the resulting protein nonfunctional. Such mutations are responsible for diseases such as Epidermolysis bullosa, sickle-cell disease, and SOD1-mediated ALS.[49] On the other hand, if a missense mutation occurs in an amino acid codon that results in the use of a different, but chemically similar, amino acid, then sometimes little or no change is rendered in the protein. For example, a change from AAA to AGA will encode arginine, a chemically similar molecule to the intended lysine. In this latter case the mutation will have little or no effect on phenotype and therefore be neutral.
- A nonsense mutation is a point mutation in a sequence of DNA that results in a premature stop codon, or a nonsense codon in the transcribed mRNA, and possibly a truncated, and often nonfunctional protein product. This sort of mutation has been linked to different diseases, such as congenital adrenal hyperplasia. (See Stop codon.)
By effect on function[edit]
A mutation becomes an effect on function mutation when the exactitude of functions between a mutated protein and its direct interactor undergoes change. The interactors can be other proteins, molecules, nucleic acids, etc. There are many mutations that fall under the category of by effect on function, but depending on the specificity of the change the mutations listed below will occur.[50]
- Loss-of-function mutations, also called inactivating mutations, result in the gene product having less or no function (being partially or wholly inactivated). When the allele has a complete loss of function (null allele), it is often called an amorph or amorphic mutation in Muller’s morphs schema. Phenotypes associated with such mutations are most often recessive. Exceptions are when the organism is haploid, or when the reduced dosage of a normal gene product is not enough for a normal phenotype (this is called haploinsufficiency). A disease that is caused by a loss-of-function mutation is Gitelman syndrome and cystic fibrosis.[51]
- Gain-of-function mutations also called activating mutations, change the gene product such that its effect gets stronger (enhanced activation) or even is superseded by a different and abnormal function. When the new allele is created, a heterozygote containing the newly created allele as well as the original will express the new allele; genetically this defines the mutations as dominant phenotypes. Several of Muller’s morphs correspond to the gain of function, including hypermorph (increased gene expression) and neomorph (novel function).
- Dominant negative mutations (also called anti-morphic mutations) have an altered gene product that acts antagonistically to the wild-type allele. These mutations usually result in an altered molecular function (often inactive) and are characterized by a dominant or semi-dominant phenotype. In humans, dominant negative mutations have been implicated in cancer (e.g., mutations in genes p53, ATM, CEBPA, and PPARgamma). Marfan syndrome is caused by mutations in the FBN1 gene, located on chromosome 15, which encodes fibrillin-1, a glycoprotein component of the extracellular matrix. Marfan syndrome is also an example of dominant negative mutation and haploinsufficiency.
- Lethal mutations result in the instant death of the developing organism. Lethal mutations can also lead to a substantial loss in the life expectancy of the organism. An example of a disease that is caused by a dominant lethal mutation is Huntington’s disease.
- Null mutations, also known as Amorphic mutations, are a form of loss-of-function mutations that completely prohibit the gene’s function. The mutation leads to a complete loss of operation at the phenotypic level, also causing no gene product to be formed. Atopic eczema and dermatitis syndrome are common diseases caused by a null mutation of the gene that activates filaggrin.
- Suppressor mutations are a type of mutation that causes the double mutation to appear normally. In suppressor mutations the phenotypic activity of a different mutation is completely suppressed, thus causing the double mutation to look normal. There are two types of suppressor mutations, there are intragenic and extragenic suppressor mutations. Intragenic mutations occur in the gene where the first mutation occurs, while extragenic mutations occur in the gene that interacts with the product of the first mutation. A common disease that results from this type of mutation is Alzheimer’s disease.[52]
- Neomorphic mutations are a part of the gain-of-function mutations and are characterized by the control of new protein product synthesis. The newly synthesized gene normally contains a novel gene expression or molecular function. The result of the neomorphic mutation is the gene where the mutation occurs has a complete change in function.[53]
- A back mutation or reversion is a point mutation that restores the original sequence and hence the original phenotype.[54]
By effect on fitness (harmful, beneficial, neutral mutations)[edit]
In genetics, it is sometimes useful to classify mutations as either harmful or beneficial (or neutral):
- A harmful, or deleterious, mutation decreases the fitness of the organism. Many, but not all mutations in essential genes are harmful (if a mutation does not change the amino acid sequence in an essential protein, it is harmless in most cases).
- A beneficial, or advantageous mutation increases the fitness of the organism. Examples are mutations that lead to antibiotic resistance in bacteria (which are beneficial for bacteria but usually not for humans).
- A neutral mutation has no harmful or beneficial effect on the organism. Such mutations occur at a steady rate, forming the basis for the molecular clock. In the neutral theory of molecular evolution, neutral mutations provide genetic drift as the basis for most variation at the molecular level. In animals or plants, most mutations are neutral, given that the vast majority of their genomes is either non-coding or consists of repetitive sequences that have no obvious function («junk DNA»).[55]
Large-scale quantitative mutagenesis screens, in which thousands of millions of mutations are tested, invariably find that a larger fraction of mutations has harmful effects but always returns a number of beneficial mutations as well. For instance, in a screen of all gene deletions in E. coli, 80% of mutations were negative, but 20% were positive, even though many had a very small effect on growth (depending on condition).[56] Gene deletions involve removal of whole genes, so that point mutations almost always have a much smaller effect. In a similar screen in Streptococcus pneumoniae, but this time with transposon insertions, 76% of insertion mutants were classified as neutral, 16% had a significantly reduced fitness, but 6% were advantageous.[57]
This classification is obviously relative and somewhat artificial: a harmful mutation can quickly turn into a beneficial mutations when conditions change. Also, there is a gradient from harmful/beneficial to neutral, as many mutations may have small and mostly neglectable effects but under certain conditions will become relevant. Also, many traits are determined by hundreds of genes (or loci), so that each locus has only a minor effect. For instance, human height is determined by hundreds of genetic variants («mutations») but each of them has a very minor effect on height,[58] apart from the impact of nutrition. Height (or size) itself may be more or less beneficial as the huge range of sizes in animal or plant groups shows.
Distribution of fitness effects (DFE)[edit]
Attempts have been made to infer the distribution of fitness effects (DFE) using mutagenesis experiments and theoretical models applied to molecular sequence data. DFE, as used to determine the relative abundance of different types of mutations (i.e., strongly deleterious, nearly neutral or advantageous), is relevant to many evolutionary questions, such as the maintenance of genetic variation,[59] the rate of genomic decay,[60] the maintenance of outcrossing sexual reproduction as opposed to inbreeding[61] and the evolution of sex and genetic recombination.[62] DFE can also be tracked by tracking the skewness of the distribution of mutations with putatively severe effects as compared to the distribution of mutations with putatively mild or absent effect.[63] In summary, the DFE plays an important role in predicting evolutionary dynamics.[64][65] A variety of approaches have been used to study the DFE, including theoretical, experimental and analytical methods.
- Mutagenesis experiment: The direct method to investigate the DFE is to induce mutations and then measure the mutational fitness effects, which has already been done in viruses, bacteria, yeast, and Drosophila. For example, most studies of the DFE in viruses used site-directed mutagenesis to create point mutations and measure relative fitness of each mutant.[66][67][68][69] In Escherichia coli, one study used transposon mutagenesis to directly measure the fitness of a random insertion of a derivative of Tn10.[70] In yeast, a combined mutagenesis and deep sequencing approach has been developed to generate high-quality systematic mutant libraries and measure fitness in high throughput.[71] However, given that many mutations have effects too small to be detected[72] and that mutagenesis experiments can detect only mutations of moderately large effect; DNA sequence analysis can provide valuable information about these mutations.
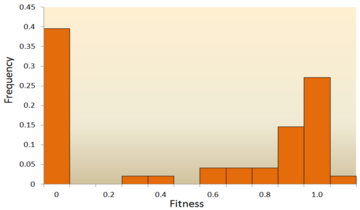
The distribution of fitness effects (DFE) of mutations in vesicular stomatitis virus. In this experiment, random mutations were introduced into the virus by site-directed mutagenesis, and the fitness of each mutant was compared with the ancestral type. A fitness of zero, less than one, one, more than one, respectively, indicates that mutations are lethal, deleterious, neutral, and advantageous.[66]
-

This figure shows a simplified version of loss-of-function, switch-of-function, gain-of-function, and conservation-of-function mutations.
Molecular sequence analysis: With rapid development of DNA sequencing technology, an enormous amount of DNA sequence data is available and even more is forthcoming in the future. Various methods have been developed to infer the DFE from DNA sequence data.[73][74][75][76] By examining DNA sequence differences within and between species, we are able to infer various characteristics of the DFE for neutral, deleterious and advantageous mutations.[24] To be specific, the DNA sequence analysis approach allows us to estimate the effects of mutations with very small effects, which are hardly detectable through mutagenesis experiments.

One of the earliest theoretical studies of the distribution of fitness effects was done by Motoo Kimura, an influential theoretical population geneticist. His neutral theory of molecular evolution proposes that most novel mutations will be highly deleterious, with a small fraction being neutral.[25][77] A later proposal by Hiroshi Akashi proposed a bimodal model for the DFE, with modes centered around highly deleterious and neutral mutations.[78] Both theories agree that the vast majority of novel mutations are neutral or deleterious and that advantageous mutations are rare, which has been supported by experimental results. One example is a study done on the DFE of random mutations in vesicular stomatitis virus.[66] Out of all mutations, 39.6% were lethal, 31.2% were non-lethal deleterious, and 27.1% were neutral. Another example comes from a high throughput mutagenesis experiment with yeast.[71] In this experiment it was shown that the overall DFE is bimodal, with a cluster of neutral mutations, and a broad distribution of deleterious mutations.
Though relatively few mutations are advantageous, those that are play an important role in evolutionary changes.[79] Like neutral mutations, weakly selected advantageous mutations can be lost due to random genetic drift, but strongly selected advantageous mutations are more likely to be fixed. Knowing the DFE of advantageous mutations may lead to increased ability to predict the evolutionary dynamics. Theoretical work on the DFE for advantageous mutations has been done by John H. Gillespie[80] and H. Allen Orr.[81] They proposed that the distribution for advantageous mutations should be exponential under a wide range of conditions, which, in general, has been supported by experimental studies, at least for strongly selected advantageous mutations.[82][83][84]
In general, it is accepted that the majority of mutations are neutral or deleterious, with advantageous mutations being rare; however, the proportion of types of mutations varies between species. This indicates two important points: first, the proportion of effectively neutral mutations is likely to vary between species, resulting from dependence on effective population size; second, the average effect of deleterious mutations varies dramatically between species.[24] In addition, the DFE also differs between coding regions and noncoding regions, with the DFE of noncoding DNA containing more weakly selected mutations.[24]
By inheritance[edit]

A mutation has caused this moss rose plant to produce flowers of different colors. This is a somatic mutation that may also be passed on in the germline.
In multicellular organisms with dedicated reproductive cells, mutations can be subdivided into germline mutations, which can be passed on to descendants through their reproductive cells, and somatic mutations (also called acquired mutations),[85] which involve cells outside the dedicated reproductive group and which are not usually transmitted to descendants.
Diploid organisms (e.g., humans) contain two copies of each gene—a paternal and a maternal allele. Based on the occurrence of mutation on each chromosome, we may classify mutations into three types. A wild type or homozygous non-mutated organism is one in which neither allele is mutated.
- A heterozygous mutation is a mutation of only one allele.
- A homozygous mutation is an identical mutation of both the paternal and maternal alleles.
- Compound heterozygous mutations or a genetic compound consists of two different mutations in the paternal and maternal alleles.[86]
Germline mutation[edit]
A germline mutation in the reproductive cells of an individual gives rise to a constitutional mutation in the offspring, that is, a mutation that is present in every cell. A constitutional mutation can also occur very soon after fertilisation, or continue from a previous constitutional mutation in a parent.[87] A germline mutation can be passed down through subsequent generations of organisms.
The distinction between germline and somatic mutations is important in animals that have a dedicated germline to produce reproductive cells. However, it is of little value in understanding the effects of mutations in plants, which lack a dedicated germline. The distinction is also blurred in those animals that reproduce asexually through mechanisms such as budding, because the cells that give rise to the daughter organisms also give rise to that organism’s germline.
A new germline mutation not inherited from either parent is called a de novo mutation.
Somatic mutation[edit]
A change in the genetic structure that is not inherited from a parent, and also not passed to offspring, is called a somatic mutation.[85] Somatic mutations are not inherited by an organism’s offspring because they do not affect the germline. However, they are passed down to all the progeny of a mutated cell within the same organism during mitosis. A major section of an organism therefore might carry the same mutation. These types of mutations are usually prompted by environmental causes, such as ultraviolet radiation or any exposure to certain harmful chemicals, and can cause diseases including cancer.[88]
With plants, some somatic mutations can be propagated without the need for seed production, for example, by grafting and stem cuttings. These type of mutation have led to new types of fruits, such as the «Delicious» apple and the «Washington» navel orange.[89]
Human and mouse somatic cells have a mutation rate more than ten times higher than the germline mutation rate for both species; mice have a higher rate of both somatic and germline mutations per cell division than humans. The disparity in mutation rate between the germline and somatic tissues likely reflects the greater importance of genome maintenance in the germline than in the soma.[90]
Special classes[edit]
- Conditional mutation is a mutation that has wild-type (or less severe) phenotype under certain «permissive» environmental conditions and a mutant phenotype under certain «restrictive» conditions. For example, a temperature-sensitive mutation can cause cell death at high temperature (restrictive condition), but might have no deleterious consequences at a lower temperature (permissive condition).[91] These mutations are non-autonomous, as their manifestation depends upon presence of certain conditions, as opposed to other mutations which appear autonomously.[92] The permissive conditions may be temperature,[93] certain chemicals,[94] light[94] or mutations in other parts of the genome.[92] In vivo mechanisms like transcriptional switches can create conditional mutations. For instance, association of Steroid Binding Domain can create a transcriptional switch that can change the expression of a gene based on the presence of a steroid ligand.[95] Conditional mutations have applications in research as they allow control over gene expression. This is especially useful studying diseases in adults by allowing expression after a certain period of growth, thus eliminating the deleterious effect of gene expression seen during stages of development in model organisms.[94] DNA Recombinase systems like Cre-Lox recombination used in association with promoters that are activated under certain conditions can generate conditional mutations. Dual Recombinase technology can be used to induce multiple conditional mutations to study the diseases which manifest as a result of simultaneous mutations in multiple genes.[94] Certain inteins have been identified which splice only at certain permissive temperatures, leading to improper protein synthesis and thus, loss-of-function mutations at other temperatures.[96] Conditional mutations may also be used in genetic studies associated with ageing, as the expression can be changed after a certain time period in the organism’s lifespan.[93]
- Replication timing quantitative trait loci affects DNA replication.
Nomenclature[edit]
In order to categorize a mutation as such, the «normal» sequence must be obtained from the DNA of a «normal» or «healthy» organism (as opposed to a «mutant» or «sick» one), it should be identified and reported; ideally, it should be made publicly available for a straightforward nucleotide-by-nucleotide comparison, and agreed upon by the scientific community or by a group of expert geneticists and biologists, who have the responsibility of establishing the standard or so-called «consensus» sequence. This step requires a tremendous scientific effort. Once the consensus sequence is known, the mutations in a genome can be pinpointed, described, and classified. The committee of the Human Genome Variation Society (HGVS) has developed the standard human sequence variant nomenclature,[97] which should be used by researchers and DNA diagnostic centers to generate unambiguous mutation descriptions. In principle, this nomenclature can also be used to describe mutations in other organisms. The nomenclature specifies the type of mutation and base or amino acid changes.
- Nucleotide substitution (e.g., 76A>T) – The number is the position of the nucleotide from the 5′ end; the first letter represents the wild-type nucleotide, and the second letter represents the nucleotide that replaced the wild type. In the given example, the adenine at the 76th position was replaced by a thymine.
- If it becomes necessary to differentiate between mutations in genomic DNA, mitochondrial DNA, and RNA, a simple convention is used. For example, if the 100th base of a nucleotide sequence mutated from G to C, then it would be written as g.100G>C if the mutation occurred in genomic DNA, m.100G>C if the mutation occurred in mitochondrial DNA, or r.100g>c if the mutation occurred in RNA. Note that, for mutations in RNA, the nucleotide code is written in lower case.
- Amino acid substitution (e.g., D111E) – The first letter is the one letter code of the wild-type amino acid, the number is the position of the amino acid from the N-terminus, and the second letter is the one letter code of the amino acid present in the mutation. Nonsense mutations are represented with an X for the second amino acid (e.g. D111X).
- Amino acid deletion (e.g., ΔF508) – The Greek letter Δ (delta) indicates a deletion. The letter refers to the amino acid present in the wild type and the number is the position from the N terminus of the amino acid were it to be present as in the wild type.
Mutation rates[edit]
Mutation rates vary substantially across species, and the evolutionary forces that generally determine mutation are the subject of ongoing investigation.
In humans, the mutation rate is about 50–90 de novo mutations per genome per generation, that is, each human accumulates about 50–90 novel mutations that were not present in his or her parents. This number has been established by sequencing thousands of human trios, that is, two parents and at least one child.[98]
The genomes of RNA viruses are based on RNA rather than DNA. The RNA viral genome can be double-stranded (as in DNA) or single-stranded. In some of these viruses (such as the single-stranded human immunodeficiency virus), replication occurs quickly, and there are no mechanisms to check the genome for accuracy. This error-prone process often results in mutations.
Randomness of mutations[edit]
There is a widespread assumption that mutations are (entirely) «random» with respect to their consequences (in terms of probability). This was shown to be wrong as mutation frequency can vary across regions of the genome, with such DNA repair- and mutation-biases being associated with various factors. For instance, biologically important regions were found to be protected from mutations and mutations beneficial to the studied plant were found to be more likely – i.e. mutation is «non-random in a way that benefits the plant».[99][100]
Disease causation[edit]
Changes in DNA caused by mutation in a coding region of DNA can cause errors in protein sequence that may result in partially or completely non-functional proteins. Each cell, in order to function correctly, depends on thousands of proteins to function in the right places at the right times. When a mutation alters a protein that plays a critical role in the body, a medical condition can result. One study on the comparison of genes between different species of Drosophila suggests that if a mutation does change a protein, the mutation will most likely be harmful, with an estimated 70 percent of amino acid polymorphisms having damaging effects, and the remainder being either neutral or weakly beneficial.[8] Some mutations alter a gene’s DNA base sequence but do not change the protein made by the gene. Studies have shown that only 7% of point mutations in noncoding DNA of yeast are deleterious and 12% in coding DNA are deleterious. The rest of the mutations are either neutral or slightly beneficial.[101]
Inherited disorders[edit]
If a mutation is present in a germ cell, it can give rise to offspring that carries the mutation in all of its cells. This is the case in hereditary diseases. In particular, if there is a mutation in a DNA repair gene within a germ cell, humans carrying such germline mutations may have an increased risk of cancer. A list of 34 such germline mutations is given in the article DNA repair-deficiency disorder. An example of one is albinism, a mutation that occurs in the OCA1 or OCA2 gene. Individuals with this disorder are more prone to many types of cancers, other disorders and have impaired vision.
DNA damage can cause an error when the DNA is replicated, and this error of replication can cause a gene mutation that, in turn, could cause a genetic disorder. DNA damages are repaired by the DNA repair system of the cell. Each cell has a number of pathways through which enzymes recognize and repair damages in DNA. Because DNA can be damaged in many ways, the process of DNA repair is an important way in which the body protects itself from disease. Once DNA damage has given rise to a mutation, the mutation cannot be repaired.
Role in carcinogenesis[edit]
On the other hand, a mutation may occur in a somatic cell of an organism. Such mutations will be present in all descendants of this cell within the same organism. The accumulation of certain mutations over generations of somatic cells is part of cause of malignant transformation, from normal cell to cancer cell.[102]
Cells with heterozygous loss-of-function mutations (one good copy of gene and one mutated copy) may function normally with the unmutated copy until the good copy has been spontaneously somatically mutated. This kind of mutation happens often in living organisms, but it is difficult to measure the rate. Measuring this rate is important in predicting the rate at which people may develop cancer.[103]
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
Prion mutations[edit]
Prions are proteins and do not contain genetic material. However, prion replication has been shown to be subject to mutation and natural selection just like other forms of replication.[104] The human gene PRNP codes for the major prion protein, PrP, and is subject to mutations that can give rise to disease-causing prions.
Beneficial mutations[edit]
Although mutations that cause changes in protein sequences can be harmful to an organism, on occasions the effect may be positive in a given environment. In this case, the mutation may enable the mutant organism to withstand particular environmental stresses better than wild-type organisms, or reproduce more quickly. In these cases a mutation will tend to become more common in a population through natural selection. Examples include the following:
HIV resistance: a specific 32 base pair deletion in human CCR5 (CCR5-Δ32) confers HIV resistance to homozygotes and delays AIDS onset in heterozygotes.[105] One possible explanation of the etiology of the relatively high frequency of CCR5-Δ32 in the European population is that it conferred resistance to the bubonic plague in mid-14th century Europe. People with this mutation were more likely to survive infection; thus its frequency in the population increased.[106] This theory could explain why this mutation is not found in Southern Africa, which remained untouched by bubonic plague. A newer theory suggests that the selective pressure on the CCR5 Delta 32 mutation was caused by smallpox instead of the bubonic plague.[107]
Malaria resistance: An example of a harmful mutation is sickle-cell disease, a blood disorder in which the body produces an abnormal type of the oxygen-carrying substance hemoglobin in the red blood cells. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the allele, because, in areas where malaria is common, there is a survival value in carrying only a single sickle-cell allele (sickle cell trait).[108] Those with only one of the two alleles of the sickle-cell disease are more resistant to malaria, since the infestation of the malaria Plasmodium is halted by the sickling of the cells that it infests.
Antibiotic resistance: Practically all bacteria develop antibiotic resistance when exposed to antibiotics. In fact, bacterial populations already have such mutations that get selected under antibiotic selection.[109] Obviously, such mutations are only beneficial for the bacteria but not for those infected.
Lactase persistence. A mutation allowed humans to express the enzyme lactase after they are naturally weaned from breast milk, allowing adults to digest lactose, which is likely one of the most beneficial mutations in recent human evolution.[110]
Compensated pathogenic deviations[edit]
Compensated pathogenic deviations refer to amino acid residues in a protein sequence that are pathogenic in one species but are wild type residues in the functionally equivalent protein in another species. Although the amino acid residue is pathogenic in the first species, it is not so in the second species because its pathogenicity is compensated by one or more amino acid substitutions in the second species. The compensatory mutation can occur in the same protein or in another protein with which it interacts.[111]
It is critical to understand the effects of compensatory mutations in the context of fixed deleterious mutations due to the population fitness decreasing because of fixation.[112] Effective population size refers to a population that is reproducing.[113] An increase in this population size has been correlated with a decreased rate of genetic diversity.[113] The position of a population relative to the critical effect population size is essential to determine the effect deleterious alleles will have on fitness.[112] If the population is below the critical effective size fitness will decrease drastically, however if the population is above the critical effect size, fitness can increase regardless of deleterious mutations due to compensatory alleles.[112]
Compensatory mutations in RNA[edit]
As the function of a RNA molecule is dependent on its structure,[114] the structure of RNA molecules is evolutionarily conserved. Therefore, any mutation that alters the stable structure of RNA molecules must be compensated by other compensatory mutations. In the context of RNA, the sequence of the RNA can be considered as ‘ genotype’ and the structure of the RNA can be considered as its ‘phenotype’. Since RNAs have relatively simpler composition than proteins, the structure of RNA molecules can be computationally predicted with high degree of accuracy. Because of this convenience, compensatory mutations have been studied in computational simulations using RNA folding algorithms.[115][116]
Evolutionary mechanism of compensation[edit]
Compensatory mutations can be explained by the genetic phenomenon epistasis whereby the phenotypic effect of one mutation is dependent upon mutation(s) at other loci. While epistasis was originally conceived in the context of interaction between different genes, intragenic epistasis has also been studied recently.[117] Existence of compensated pathogenic deviations can be explained by ‘sign epistasis’, in which the effects of a deleterious mutation can be compensated by the presence of a epistatic mutation in another loci. For a given protein, a deleterious mutation (D) and a compensatory mutation (C) can be considered, where C can be in the same protein as D or in a different interacting protein depending on the context. The fitness effect of C itself could be neutral or somewhat deleterious such that it can still exist in the population, and the effect of D is deleterious to the extent that it cannot exist in the population. However, when C and D co-occur together, the combined fitness effect becomes neutral or positive.[111] Thus, compensatory mutations can bring novelty to proteins by forging new pathways of protein evolution : it allows individuals to travel from one fitness peak to another through the valleys of lower fitness.[117]
DePristo et al. 2005 outlined two models to explain the dynamics of compensatory pathogenic deviations (CPD).[118] In the first hypothesis P is a pathogenic amino acid mutation that and C is a neutral compensatory mutation.[118] Under these conditions, if the pathogenic mutation arises after a compensatory mutation, then P can become fixed in the population.[118] The second model of CPDs states that P and C are both deleterious mutations resulting in fitness valleys when mutations occur simultaneously.[118] Using publicly available, Ferrer-Costa et al. 2007 obtained compensatory mutations and human pathogenic mutation datasets that were characterized to determine what causes CPDs.[119] Results indicate that the structural constraints and the location in protein structure determine whether compensated mutations will occur.[119]
Experimental evidence of compensatory mutations[edit]
Experiment in bacteria[edit]
Lunzer et al.[120] tested the outcome of swapping divergent amino acids between two orthologous proteins of isopropymalate dehydrogenase (IMDH). They substituted 168 amino acids in Escherichia coli IMDH that are wild type residues in IMDH Pseudomonas aeruginosa. They found that over one third of these substitutions compromised IMDH enzymatic activity in the Escherichia coli genetic background. This demonstrated that identical amino acid states can result in different phenotypic states depending on the genetic background. Corrigan et al. 2011 demonstrated how staphylococcus aureus was able to grow normally without the presence of lipoteichoic acid due to compensatory mutations.[121] Whole genome sequencing results revealed that when Cyclic-di-AMP phosphodiesterase (GdpP) was disrupted in this bacterium, it compensated for the disappearance of the cell wall polymer, resulting in normal cell growth.[121]
Research has shown that bacteria can gain drug resistance through compensatory mutations that do not impede or having little effect on fitness.[122] Previous research from Gagneux et al. 2006 has found that laboratory grown M. tuberculosis strains with rifampicin resistance have reduced fitness, however drug resistant clinical strains of this pathogenic bacteria do not have reduced fitness.[123] Comas et al. 2012 used whole genome comparisons between clinical strains and lab derived mutants to determine the role and contribution of compensatory mutations in drug resistance to rifampicin.[122] Genome analysis reveal rifampicin resistant strains have a mutation in rpoA and rpoC.[122] A similar study investigated the bacterial fitness associated with compensatory mutations in rifampin resistant Escherichia coli.[124] Results obtained from this study demonstrate that drug resistance is linked to bacterial fitness as higher fitness costs are linked to greater transcription errors.[124]
Experiment in virus[edit]
Gong et al.[125] collected obtained genotype data of influenza nucleoprotein from different timelines and temporally ordered them according to their time of origin. Then they isolated 39 amino acid substitutions that occurred in different timelines and substituted them in a genetic background that approximated the ancestral genotype. They found that 3 of the 39 substitutions significantly reduced the fitness of the ancestral background. Compensatory mutations are new mutations that arise and have a positive or neutral impact on a populations fitness.[126] Previous research has shown that populations have can compensate detrimental mutations.[111][126][127] Burch and Chao tested Fisher’s geometric model of adaptive evolution by testing whether bacteriophage φ6 evolves by small steps.[128] Their results showed that bacteriophage φ6 fitness declined rapidly and recovered in small steps .[128] Viral nucleoproteins have been shown to avoid cytotoxic T lymphocytes (CTLs) through arginine-to glycine substitutions.[129] This substitution mutations impacts the fitness of viral nucleoproteins, however compensatory co-mutations impede fitness declines and aid the virus to avoid recognition from CTLs.[129] Mutations can have three different effects; mutations can have deleterious effects, some increase fitness through compensatory mutations, and lastly mutations can be counterbalancing resulting in compensatory neutral mutations.[130][124][123]
See also[edit]
- Aneuploidy
- Antioxidant
- Budgerigar colour genetics
- DbDNV (2010)
- Deletion (genetics)
- Ecogenetics
- Embryology
- Homeobox
- Human somatic variation
- Polyploidy
- Robertsonian translocation
- Signature-tagged mutagenesis
- Somatic hypermutation
- TILLING (molecular biology)
- Trinucleotide repeat expansion
References[edit]
- ^ «mutation | Learn Science at Scitable». Nature. Nature Education. Retrieved 24 September 2018.
- ^ Sfeir A, Symington LS (November 2015). «Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway?». Trends in Biochemical Sciences. 40 (11): 701–714. doi:10.1016/j.tibs.2015.08.006. PMC 4638128. PMID 26439531.
- ^ Chen J, Miller BF, Furano AV (April 2014). «Repair of naturally occurring mismatches can induce mutations in flanking DNA». eLife. 3: e02001. doi:10.7554/elife.02001. PMC 3999860. PMID 24843013.
- ^ Rodgers K, McVey M (January 2016). «Error-Prone Repair of DNA Double-Strand Breaks». Journal of Cellular Physiology. 231 (1): 15–24. doi:10.1002/jcp.25053. PMC 4586358. PMID 26033759.
- ^ a b Bertram JS (December 2000). «The molecular biology of cancer». Molecular Aspects of Medicine. 21 (6): 167–223. doi:10.1016/S0098-2997(00)00007-8. PMID 11173079. S2CID 24155688.
- ^ a b Aminetzach YT, Macpherson JM, Petrov DA (July 2005). «Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila». Science. 309 (5735): 764–7. Bibcode:2005Sci…309..764A. doi:10.1126/science.1112699. PMID 16051794. S2CID 11640993.
- ^ Burrus V, Waldor MK (June 2004). «Shaping bacterial genomes with integrative and conjugative elements». Research in Microbiology. 155 (5): 376–86. doi:10.1016/j.resmic.2004.01.012. PMID 15207870.
- ^ a b Sawyer SA, Parsch J, Zhang Z, Hartl DL (April 2007). «Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila». Proceedings of the National Academy of Sciences of the United States of America. 104 (16): 6504–10. Bibcode:2007PNAS..104.6504S. doi:10.1073/pnas.0701572104. PMC 1871816. PMID 17409186.
- ^ Hastings PJ, Lupski JR, Rosenberg SM, Ira G (August 2009). «Mechanisms of change in gene copy number». Nature Reviews. Genetics. 10 (8): 551–64. doi:10.1038/nrg2593. PMC 2864001. PMID 19597530.
- ^ Carroll SB, Grenier JK, Weatherbee SD (2005). From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design (2nd ed.). Malden, MA: Blackwell Publishing. ISBN 978-1-4051-1950-4. LCCN 2003027991. OCLC 53972564.
- ^ Harrison PM, Gerstein M (May 2002). «Studying genomes through the aeons: protein families, pseudogenes and proteome evolution». Journal of Molecular Biology. 318 (5): 1155–74. doi:10.1016/S0022-2836(02)00109-2. PMID 12083509.
- ^ Orengo CA, Thornton JM (July 2005). «Protein families and their evolution-a structural perspective». Annual Review of Biochemistry. 74: 867–900. doi:10.1146/annurev.biochem.74.082803.133029. PMID 15954844.
- ^ Long M, Betrán E, Thornton K, Wang W (November 2003). «The origin of new genes: glimpses from the young and old». Nature Reviews. Genetics. 4 (11): 865–75. doi:10.1038/nrg1204. PMID 14634634. S2CID 33999892.
- ^ Wang M, Caetano-Anollés G (January 2009). «The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world». Structure. 17 (1): 66–78. doi:10.1016/j.str.2008.11.008. PMID 19141283.
- ^ Bowmaker JK (May 1998). «Evolution of colour vision in vertebrates». Eye. 12 (Pt 3b): 541–7. doi:10.1038/eye.1998.143. PMID 9775215. S2CID 12851209.
- ^ Gregory TR, Hebert PD (April 1999). «The modulation of DNA content: proximate causes and ultimate consequences». Genome Research. 9 (4): 317–24. doi:10.1101/gr.9.4.317. PMID 10207154. S2CID 16791399.
- ^ Hurles M (July 2004). «Gene duplication: the genomic trade in spare parts». PLOS Biology. 2 (7): E206. doi:10.1371/journal.pbio.0020206. PMC 449868. PMID 15252449.
- ^ Liu N, Okamura K, Tyler DM, Phillips MD, Chung WJ, Lai EC (October 2008). «The evolution and functional diversification of animal microRNA genes». Cell Research. 18 (10): 985–96. doi:10.1038/cr.2008.278. PMC 2712117. PMID 18711447.
- ^ Siepel A (October 2009). «Darwinian alchemy: Human genes from noncoding DNA». Genome Research. 19 (10): 1693–5. doi:10.1101/gr.098376.109. PMC 2765273. PMID 19797681.
- ^ Zhang J, Wang X, Podlaha O (May 2004). «Testing the chromosomal speciation hypothesis for humans and chimpanzees». Genome Research. 14 (5): 845–51. doi:10.1101/gr.1891104. PMC 479111. PMID 15123584.
- ^ Ayala FJ, Coluzzi M (May 2005). «Chromosome speciation: humans, Drosophila, and mosquitoes». Proceedings of the National Academy of Sciences of the United States of America. 102 (Suppl 1): 6535–42. Bibcode:2005PNAS..102.6535A. doi:10.1073/pnas.0501847102. PMC 1131864. PMID 15851677.
- ^ Hurst GD, Werren JH (August 2001). «The role of selfish genetic elements in eukaryotic evolution». Nature Reviews Genetics. 2 (8): 597–606. doi:10.1038/35084545. PMID 11483984. S2CID 2715605.
- ^ Häsler J, Strub K (November 2006). «Alu elements as regulators of gene expression». Nucleic Acids Research. 34 (19): 5491–7. doi:10.1093/nar/gkl706. PMC 1636486. PMID 17020921.
- ^ a b c d Eyre-Walker A, Keightley PD (August 2007). «The distribution of fitness effects of new mutations» (PDF). Nature Reviews Genetics. 8 (8): 610–8. doi:10.1038/nrg2146. PMID 17637733. S2CID 10868777. Archived from the original (PDF) on 4 March 2016. Retrieved 6 September 2010.
- ^ a b Kimura M (1983). The Neutral Theory of Molecular Evolution. Cambridge, UK; New York: Cambridge University Press. ISBN 978-0-521-23109-1. LCCN 82022225. OCLC 9081989.
- ^ Bohidar HB (January 2015). Fundamentals of Polymer Physics and Molecular Biophysics. Cambridge University Press. ISBN 978-1-316-09302-3.
- ^ Dover GA, Darwin C (2000). Dear Mr. Darwin: Letters on the Evolution of Life and Human Nature. University of California Press. ISBN 9780520227903.
- ^ Tibayrenc M (12 January 2017). Genetics and Evolution of Infectious Diseases. Elsevier. ISBN 9780128001530.
- ^ «Cancer Is Partly Caused By Bad Luck, Study Finds». NPR.org. Archived from the original on 13 July 2017.
- ^ Jha A (22 August 2012). «Older fathers pass on more genetic mutations, study shows». The Guardian.
- ^ Ames BN, Shigenaga MK, Hagen TM (September 1993). «Oxidants, antioxidants, and the degenerative diseases of aging». Proceedings of the National Academy of Sciences of the United States of America. 90 (17): 7915–22. Bibcode:1993PNAS…90.7915A. doi:10.1073/pnas.90.17.7915. PMC 47258. PMID 8367443.
- ^ Montelone BA (1998). «Mutation, Mutagens, and DNA Repair». www-personal.ksu.edu. Archived from the original on 26 September 2015. Retrieved 2 October 2015.
- ^ Slocombe L, Al-Khalili JS, Sacchi M (February 2021). «Quantum and classical effects in DNA point mutations: Watson-Crick tautomerism in AT and GC base pairs». Physical Chemistry Chemical Physics. 23 (7): 4141–4150. Bibcode:2021PCCP…23.4141S. doi:10.1039/D0CP05781A. ISSN 1463-9076. PMID 33533770. S2CID 231788542.
- ^ Slocombe L, Sacchi M, Al-Khalili J (5 May 2022). «An open quantum systems approach to proton tunnelling in DNA». Communications Physics. 5 (1): 109. arXiv:2110.00113. Bibcode:2022CmPhy…5..109S. doi:10.1038/s42005-022-00881-8. ISSN 2399-3650. S2CID 238253421.
- ^ Stuart GR, Oda Y, de Boer JG, Glickman BW (March 2000). «Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice». Genetics. 154 (3): 1291–300. doi:10.1093/genetics/154.3.1291. PMC 1460990. PMID 10757770.
- ^ Kunz BA, Ramachandran K, Vonarx EJ (April 1998). «DNA sequence analysis of spontaneous mutagenesis in Saccharomyces cerevisiae». Genetics. 148 (4): 1491–505. doi:10.1093/genetics/148.4.1491. PMC 1460101. PMID 9560369.
- ^ Lieber MR (July 2010). «The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway». Annual Review of Biochemistry. 79: 181–211. doi:10.1146/annurev.biochem.052308.093131. PMC 3079308. PMID 20192759.
- ^ Created from PDB 1JDG Archived 31 December 2015 at the Wayback Machine
- ^ Pfohl-Leszkowicz A, Manderville RA (January 2007). «Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans». Molecular Nutrition & Food Research. 51 (1): 61–99. doi:10.1002/mnfr.200600137. PMID 17195275.
- ^ Kozmin S, Slezak G, Reynaud-Angelin A, Elie C, de Rycke Y, Boiteux S, Sage E (September 2005). «UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae». Proceedings of the National Academy of Sciences of the United States of America. 102 (38): 13538–43. Bibcode:2005PNAS..10213538K. doi:10.1073/pnas.0504497102. PMC 1224634. PMID 16157879.
- ^ a b Fitzgerald DM, Rosenberg SM (April 2019). «What is mutation? A chapter in the series: How microbes «jeopardize» the modern synthesis». PLOS Genetics. 15 (4): e1007995. doi:10.1371/journal.pgen.1007995. PMC 6443146. PMID 30933985.
- ^ Galhardo RS, Hastings PJ, Rosenberg SM (1 January 2007). «Mutation as a stress response and the regulation of evolvability». Critical Reviews in Biochemistry and Molecular Biology. 42 (5): 399–435. doi:10.1080/10409230701648502. PMC 3319127. PMID 17917874.
- ^ Quinto-Alemany D, Canerina-Amaro A, Hernández-Abad LG, Machín F, Romesberg FE, Gil-Lamaignere C (31 July 2012). Sturtevant J (ed.). «Yeasts acquire resistance secondary to antifungal drug treatment by adaptive mutagenesis». PLOS ONE. 7 (7): e42279. Bibcode:2012PLoSO…742279Q. doi:10.1371/journal.pone.0042279. PMC 3409178. PMID 22860105.
- ^ Rahman N. «The clinical impact of DNA sequence changes». Transforming Genetic Medicine Initiative. Archived from the original on 4 August 2017. Retrieved 27 June 2017.
- ^ Freese E (April 1959). «The Difference Between Spontaneous and Base-Analogue Induced Mutations of Phage T4». Proceedings of the National Academy of Sciences of the United States of America. 45 (4): 622–33. Bibcode:1959PNAS…45..622F. doi:10.1073/pnas.45.4.622. PMC 222607. PMID 16590424.
- ^ Freese E (June 1959). «The specific mutagenic effect of base analogues on Phage T4». Journal of Molecular Biology. 1 (2): 87–105. doi:10.1016/S0022-2836(59)80038-3.
- ^ References for the image are found in Wikimedia Commons page at: Commons:File:Notable mutations.svg#References.
- ^ Hogan CM (12 October 2010). «Mutation». In Monosson E (ed.). Encyclopedia of Earth. Washington, D.C.: Environmental Information Coalition, National Council for Science and the Environment. OCLC 72808636. Archived from the original on 14 November 2015. Retrieved 8 October 2015.
- ^ Boillée S, Vande Velde C, Cleveland DW (October 2006). «ALS: a disease of motor neurons and their nonneuronal neighbors». Neuron. 52 (1): 39–59. CiteSeerX 10.1.1.325.7514. doi:10.1016/j.neuron.2006.09.018. PMID 17015226. S2CID 12968143.
- ^ Reva B, Antipin Y, Sander C (September 2011). «Predicting the functional impact of protein mutations: application to cancer genomics». Nucleic Acids Research. 39 (17): e118. doi:10.1093/nar/gkr407. PMC 3177186. PMID 21727090.
- ^ Housden BE, Muhar M, Gemberling M, Gersbach CA, Stainier DY, Seydoux G, et al. (January 2017). «Loss-of-function genetic tools for animal models: cross-species and cross-platform differences». Nature Reviews. Genetics. 18 (1): 24–40. doi:10.1038/nrg.2016.118. PMC 5206767. PMID 27795562.
- ^ Eggertsson G, Adelberg EA (August 1965). «Map positions and specificities of suppressor mutations in Escherichia coli K-12». Genetics. 52 (2): 319–340. doi:10.1093/genetics/52.2.319. PMC 1210853. PMID 5324068.
- ^ Takiar V, Ip CK, Gao M, Mills GB, Cheung LW (March 2017). «Neomorphic mutations create therapeutic challenges in cancer». Oncogene. 36 (12): 1607–1618. doi:10.1038/onc.2016.312. PMC 6609160. PMID 27841866.
- ^ Ellis NA, Ciocci S, German J (February 2001). «Back mutation can produce phenotype reversion in Bloom syndrome somatic cells». Human Genetics. 108 (2): 167–73. doi:10.1007/s004390000447. PMID 11281456. S2CID 22290041.
- ^ Doolittle WF, Brunet TD (December 2017). «On causal roles and selected effects: our genome is mostly junk». BMC Biology. 15 (1): 116. doi:10.1186/s12915-017-0460-9. PMC 5718017. PMID 29207982.
- ^ Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, et al. (January 2011). «Phenotypic landscape of a bacterial cell». Cell. 144 (1): 143–56. doi:10.1016/j.cell.2010.11.052. PMC 3060659. PMID 21185072.
- ^ van Opijnen T, Bodi KL, Camilli A (October 2009). «Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms». Nature Methods. 6 (10): 767–72. doi:10.1038/nmeth.1377. PMC 2957483. PMID 19767758.
- ^ Allen HL, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. (October 2010). «Hundreds of variants clustered in genomic loci and biological pathways affect human height». Nature. 467 (7317): 832–8. Bibcode:2010Natur.467..832L. doi:10.1038/nature09410. PMC 2955183. PMID 20881960.
- ^ Charlesworth D, Charlesworth B, Morgan MT (December 1995). «The pattern of neutral molecular variation under the background selection model». Genetics. 141 (4): 1619–32. doi:10.1093/genetics/141.4.1619. PMC 1206892. PMID 8601499.
- ^ Loewe L (April 2006). «Quantifying the genomic decay paradox due to Muller’s ratchet in human mitochondrial DNA». Genetical Research. 87 (2): 133–59. doi:10.1017/S0016672306008123. PMID 16709275.
- ^ Bernstein H, Hopf FA, Michod RE (1987). «The molecular basis of the evolution of sex». Molecular Genetics of Development. Advances in Genetics. Vol. 24. pp. 323–70. doi:10.1016/s0065-2660(08)60012-7. ISBN 9780120176243. PMID 3324702.
- ^ Peck JR, Barreau G, Heath SC (April 1997). «Imperfect genes, Fisherian mutation and the evolution of sex». Genetics. 145 (4): 1171–99. doi:10.1093/genetics/145.4.1171. PMC 1207886. PMID 9093868.
- ^ Simcikova D, Heneberg P (December 2019). «Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases». Scientific Reports. 9 (1): 18577. Bibcode:2019NatSR…918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.
- ^ Keightley PD, Lynch M (March 2003). «Toward a realistic model of mutations affecting fitness». Evolution; International Journal of Organic Evolution. 57 (3): 683–5, discussion 686–9. doi:10.1554/0014-3820(2003)057[0683:tarmom]2.0.co;2. JSTOR 3094781. PMID 12703958. S2CID 198157678.
- ^ Barton NH, Keightley PD (January 2002). «Understanding quantitative genetic variation». Nature Reviews Genetics. 3 (1): 11–21. doi:10.1038/nrg700. PMID 11823787. S2CID 8934412.
- ^ a b c Sanjuán R, Moya A, Elena SF (June 2004). «The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus». Proceedings of the National Academy of Sciences of the United States of America. 101 (22): 8396–401. Bibcode:2004PNAS..101.8396S. doi:10.1073/pnas.0400146101. PMC 420405. PMID 15159545.
- ^ Carrasco P, de la Iglesia F, Elena SF (December 2007). «Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus». Journal of Virology. 81 (23): 12979–84. doi:10.1128/JVI.00524-07. PMC 2169111. PMID 17898073.
- ^ Sanjuán R (June 2010). «Mutational fitness effects in RNA and single-stranded DNA viruses: common patterns revealed by site-directed mutagenesis studies». Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 365 (1548): 1975–82. doi:10.1098/rstb.2010.0063. PMC 2880115. PMID 20478892.
- ^ Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R (June 2010). «Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1». Genetics. 185 (2): 603–9. doi:10.1534/genetics.110.115162. PMC 2881140. PMID 20382832.
- ^ Elena SF, Ekunwe L, Hajela N, Oden SA, Lenski RE (March 1998). «Distribution of fitness effects caused by random insertion mutations in Escherichia coli». Genetica. 102–103 (1–6): 349–58. doi:10.1023/A:1017031008316. PMID 9720287. S2CID 2267064.
- ^ a b Hietpas RT, Jensen JD, Bolon DN (May 2011). «Experimental illumination of a fitness landscape». Proceedings of the National Academy of Sciences of the United States of America. 108 (19): 7896–901. Bibcode:2011PNAS..108.7896H. doi:10.1073/pnas.1016024108. PMC 3093508. PMID 21464309.
- ^ Davies EK, Peters AD, Keightley PD (September 1999). «High frequency of cryptic deleterious mutations in Caenorhabditis elegans». Science. 285 (5434): 1748–51. doi:10.1126/science.285.5434.1748. PMID 10481013.
- ^ Loewe L, Charlesworth B (September 2006). «Inferring the distribution of mutational effects on fitness in Drosophila». Biology Letters. 2 (3): 426–30. doi:10.1098/rsbl.2006.0481. PMC 1686194. PMID 17148422.
- ^ Eyre-Walker A, Woolfit M, Phelps T (June 2006). «The distribution of fitness effects of new deleterious amino acid mutations in humans». Genetics. 173 (2): 891–900. doi:10.1534/genetics.106.057570. PMC 1526495. PMID 16547091.
- ^ Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL (August 2003). «Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection». Journal of Molecular Evolution. 57 (1): S154–64. Bibcode:2003JMolE..57S.154S. CiteSeerX 10.1.1.78.65. doi:10.1007/s00239-003-0022-3. PMID 15008412. S2CID 18051307.
- ^ Piganeau G, Eyre-Walker A (September 2003). «Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock». Proceedings of the National Academy of Sciences of the United States of America. 100 (18): 10335–40. Bibcode:2003PNAS..10010335P. doi:10.1073/pnas.1833064100. PMC 193562. PMID 12925735.
- ^ Kimura M (February 1968). «Evolutionary rate at the molecular level». Nature. 217 (5129): 624–6. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732. S2CID 4161261.
- ^ Akashi H (September 1999). «Within- and between-species DNA sequence variation and the ‘footprint’ of natural selection». Gene. 238 (1): 39–51. doi:10.1016/S0378-1119(99)00294-2. PMID 10570982.
- ^ Eyre-Walker A (October 2006). «The genomic rate of adaptive evolution». Trends in Ecology & Evolution. 21 (10): 569–75. doi:10.1016/j.tree.2006.06.015. PMID 16820244.
- ^ Gillespie JH (September 1984). «Molecular Evolution Over the Mutational Landscape». Evolution. 38 (5): 1116–1129. doi:10.2307/2408444. JSTOR 2408444. PMID 28555784.
- ^ Orr HA (April 2003). «The distribution of fitness effects among beneficial mutations». Genetics. 163 (4): 1519–26. doi:10.1093/genetics/163.4.1519. PMC 1462510. PMID 12702694.
- ^ Kassen R, Bataillon T (April 2006). «Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria». Nature Genetics. 38 (4): 484–8. doi:10.1038/ng1751. PMID 16550173. S2CID 6954765.
- ^ Rokyta DR, Joyce P, Caudle SB, Wichman HA (April 2005). «An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus». Nature Genetics. 37 (4): 441–4. doi:10.1038/ng1535. PMID 15778707. S2CID 20296781.
- ^ Imhof M, Schlotterer C (January 2001). «Fitness effects of advantageous mutations in evolving Escherichia coli populations». Proceedings of the National Academy of Sciences of the United States of America. 98 (3): 1113–7. Bibcode:2001PNAS…98.1113I. doi:10.1073/pnas.98.3.1113. PMC 14717. PMID 11158603.
- ^ a b «Somatic cell genetic mutation». Genome Dictionary. Athens, Greece: Information Technology Associates. 30 June 2007. Archived from the original on 24 February 2010. Retrieved 6 June 2010.
- ^ «Compound heterozygote». MedTerms. New York: WebMD. 14 June 2012. Archived from the original on 4 March 2016. Retrieved 9 October 2015.
- ^ «RB1 Genetics». Daisy’s Eye Cancer Fund. Oxford, UK. Archived from the original on 26 November 2011. Retrieved 9 October 2015.
- ^ «somatic mutation | genetics». Encyclopædia Britannica. Archived from the original on 31 March 2017. Retrieved 31 March 2017.
- ^ Hartl L, Jones EW (1998). Genetics Principles and Analysis. Sudbury, Massachusetts: Jones and Bartlett Publishers. pp. 556. ISBN 978-0-7637-0489-6.
- ^ Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J (May 2017). «Differences between germline and somatic mutation rates in humans and mice». Nature Communications. 8: 15183. Bibcode:2017NatCo…815183M. doi:10.1038/ncomms15183. PMC 5436103. PMID 28485371.
- ^ Alberts B (2014). Molecular Biology of the Cell (6 ed.). Garland Science. p. 487. ISBN 9780815344322.
- ^ a b Chadov BF, Fedorova NB, Chadova EV (1 July 2015). «Conditional mutations in Drosophila melanogaster: On the occasion of the 150th anniversary of G. Mendel’s report in Brünn». Mutation Research/Reviews in Mutation Research. 765: 40–55. doi:10.1016/j.mrrev.2015.06.001. PMID 26281767.
- ^ a b Landis G, Bhole D, Lu L, Tower J (July 2001). «High-frequency generation of conditional mutations affecting Drosophila melanogaster development and life span». Genetics. 158 (3): 1167–76. doi:10.1093/genetics/158.3.1167. PMC 1461716. PMID 11454765. Archived from the original on 22 March 2017. Retrieved 21 March 2017.
- ^ a b c d Gierut JJ, Jacks TE, Haigis KM (April 2014). «Strategies to achieve conditional gene mutation in mice». Cold Spring Harbor Protocols. 2014 (4): 339–49. doi:10.1101/pdb.top069807. PMC 4142476. PMID 24692485.
- ^ Spencer DM (May 1996). «Creating conditional mutations in mammals». Trends in Genetics. 12 (5): 181–7. doi:10.1016/0168-9525(96)10013-5. PMID 8984733.
- ^ Tan G, Chen M, Foote C, Tan C (September 2009). «Temperature-sensitive mutations made easy: generating conditional mutations by using temperature-sensitive inteins that function within different temperature ranges». Genetics. 183 (1): 13–22. doi:10.1534/genetics.109.104794. PMC 2746138. PMID 19596904.
- ^ den Dunnen JT, Antonarakis SE (January 2000). «Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion». Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815. S2CID 84706224.
- ^ Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, et al. (September 2017). «Parental influence on human germline de novo mutations in 1,548 trios from Iceland». Nature. 549 (7673): 519–522. Bibcode:2017Natur.549..519J. doi:10.1038/nature24018. PMID 28959963. S2CID 205260431.
- ^ «Study challenges evolutionary theory that DNA mutations are random». U.C. Davis. Retrieved 12 February 2022.
- ^ Monroe JG, Srikant T, Carbonell-Bejerano P, Becker C, Lensink M, Exposito-Alonso M, et al. (February 2022). «Mutation bias reflects natural selection in Arabidopsis thaliana». Nature. 602 (7895): 101–105. Bibcode:2022Natur.602..101M. doi:10.1038/s41586-021-04269-6. PMC 8810380. PMID 35022609.
- ^ Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC (August 2008). Pritchard JK (ed.). «A catalog of neutral and deleterious polymorphism in yeast». PLOS Genetics. 4 (8): e1000183. doi:10.1371/journal.pgen.1000183. PMC 2515631. PMID 18769710.
- ^ Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (June 1993). «Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis». Nature. 363 (6429): 558–61. Bibcode:1993Natur.363..558I. doi:10.1038/363558a0. PMID 8505985. S2CID 4254940.
- ^ Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L (September 2005). «A quantitative measurement of the human somatic mutation rate». Cancer Research. 65 (18): 8111–7. doi:10.1158/0008-5472.CAN-04-1198. PMID 16166284.
- ^ «‘Lifeless’ prion proteins are ‘capable of evolution’«. Health. BBC News. London. 1 January 2010. Archived from the original on 25 September 2015. Retrieved 10 October 2015.
- ^ Sullivan AD, Wigginton J, Kirschner D (August 2001). «The coreceptor mutation CCR5Delta32 influences the dynamics of HIV epidemics and is selected for by HIV». Proceedings of the National Academy of Sciences of the United States of America. 98 (18): 10214–9. Bibcode:2001PNAS…9810214S. doi:10.1073/pnas.181325198. PMC 56941. PMID 11517319.
- ^ «Mystery of the Black Death». Secrets of the Dead. Season 3. Episode 2. 30 October 2002. PBS. Archived from the original on 12 October 2015. Retrieved 10 October 2015. Episode background.
- ^ Galvani AP, Slatkin M (December 2003). «Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele». Proceedings of the National Academy of Sciences of the United States of America. 100 (25): 15276–9. Bibcode:2003PNAS..10015276G. doi:10.1073/pnas.2435085100. PMC 299980. PMID 14645720.
- ^ Konotey-Ahulu F. «Frequently Asked Questions [FAQ’s]». sicklecell.md. Archived from the original on 30 April 2011. Retrieved 16 April 2010.
- ^ Hughes D, Andersson DI (September 2017). «Evolutionary Trajectories to Antibiotic Resistance». Annual Review of Microbiology. 71: 579–596. doi:10.1146/annurev-micro-090816-093813. PMID 28697667.
- ^ Ségurel L, Bon C (August 2017). «On the Evolution of Lactase Persistence in Humans». Annual Review of Genomics and Human Genetics. 18: 297–319. doi:10.1146/annurev-genom-091416-035340. PMID 28426286.
- ^ a b c Barešić, Anja; Martin, Andrew C.R. (1 August 2011). «Compensated pathogenic deviations». BioMolecular Concepts. 2 (4): 281–292. doi:10.1515/bmc.2011.025. ISSN 1868-503X. PMID 25962036. S2CID 6540447.
- ^ a b c Whitlock, Michael C.; Griswold, Cortland K.; Peters, Andrew D. (2003). «Compensating for the meltdown: The critical effective size of a population with deleterious and compensatory mutations». Annales Zoologici Fennici. 40 (2): 169–183. ISSN 0003-455X. JSTOR 23736523.
- ^ a b Lanfear, Robert; Kokko, Hanna; Eyre-Walker, Adam (1 January 2014). «Population size and the rate of evolution». Trends in Ecology & Evolution. 29 (1): 33–41. doi:10.1016/j.tree.2013.09.009. ISSN 0169-5347. PMID 24148292.
- ^ Doudna, Jennifer A. (1 November 2000). «Structural genomics of RNA». Nature Structural Biology. 7: 954–956. doi:10.1038/80729. PMID 11103998. S2CID 998448.
- ^ Cowperthwaite, Matthew C.; Bull, J. J.; Meyers, Lauren Ancel (20 October 2006). «From Bad to Good: Fitness Reversals and the Ascent of Deleterious Mutations». PLOS Computational Biology. 2 (10): e141. Bibcode:2006PLSCB…2..141C. doi:10.1371/journal.pcbi.0020141. ISSN 1553-7358. PMC 1617134. PMID 17054393.
- ^ Cowperthwaite, Matthew C.; Meyers, Lauren Ancel (1 December 2007). «How Mutational Networks Shape Evolution: Lessons from RNA Models». Annual Review of Ecology, Evolution, and Systematics. 38 (1): 203–230. doi:10.1146/annurev.ecolsys.38.091206.095507. ISSN 1543-592X.
- ^ a b Azbukina, Nadezhda; Zharikova, Anastasia; Ramensky, Vasily (1 October 2022). «Intragenic compensation through the lens of deep mutational scanning». Biophysical Reviews. 14 (5): 1161–1182. doi:10.1007/s12551-022-01005-w. ISSN 1867-2469. PMC 9636336. PMID 36345285.
- ^ a b c d DePristo, Mark A.; Weinreich, Daniel M.; Hartl, Daniel L. (September 2005). «Missense meanderings in sequence space: a biophysical view of protein evolution». Nature Reviews. Genetics. 6 (9): 678–687. doi:10.1038/nrg1672. ISSN 1471-0056. PMID 16074985. S2CID 13236893.
- ^ a b Ferrer-Costa, Carles; Orozco, Modesto; Cruz, Xavier de la (5 January 2007). «Characterization of Compensated Mutations in Terms of Structural and Physico-Chemical Properties». Journal of Molecular Biology. 365 (1): 249–256. doi:10.1016/j.jmb.2006.09.053. ISSN 0022-2836. PMID 17059831.
- ^ Lunzer, Mark; Golding, G. Brian; Dean, Antony M. (21 October 2010). «Pervasive Cryptic Epistasis in Molecular Evolution». PLOS Genetics. 6 (10): e1001162. doi:10.1371/journal.pgen.1001162. ISSN 1553-7404. PMC 2958800. PMID 20975933.
- ^ a b Corrigan, Rebecca M.; Abbott, James C.; Burhenne, Heike; Kaever, Volkhard; Gründling, Angelika (1 September 2011). «c-di-AMP Is a New Second Messenger in Staphylococcus aureus with a Role in Controlling Cell Size and Envelope Stress». PLOS Pathogens. 7 (9): e1002217. doi:10.1371/journal.ppat.1002217. ISSN 1553-7366. PMC 3164647. PMID 21909268.
- ^ a b c Comas, Iñaki; Borrell, Sonia; Roetzer, Andreas; Rose, Graham; Malla, Bijaya; Kato-Maeda, Midori; Galagan, James; Niemann, Stefan; Gagneux, Sebastien (January 2012). «Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes». Nature Genetics. 44 (1): 106–110. doi:10.1038/ng.1038. ISSN 1546-1718. PMC 3246538. PMID 22179134.
- ^ a b Gagneux, Sebastien; Long, Clara Davis; Small, Peter M.; Van, Tran; Schoolnik, Gary K.; Bohannan, Brendan J. M. (30 June 2006). «The competitive cost of antibiotic resistance in Mycobacterium tuberculosis». Science. 312 (5782): 1944–1946. doi:10.1126/science.1124410. ISSN 1095-9203. PMID 16809538. S2CID 7454895.
- ^ a b c Reynolds, M. G. (December 2000). «Compensatory evolution in rifampin-resistant Escherichia coli». Genetics. 156 (4): 1471–1481. doi:10.1093/genetics/156.4.1471. ISSN 0016-6731. PMC 1461348. PMID 11102350.
- ^ Gong, Lizhi Ian; Suchard, Marc A; Bloom, Jesse D (14 May 2013). Pascual, Mercedes (ed.). «Stability-mediated epistasis constrains the evolution of an influenza protein». eLife. 2: e00631. doi:10.7554/eLife.00631. ISSN 2050-084X. PMC 3654441. PMID 23682315.
- ^ a b Davis, Brad H.; Poon, Art F.Y.; Whitlock, Michael C. (22 May 2009). «Compensatory mutations are repeatable and clustered within proteins». Proceedings of the Royal Society B: Biological Sciences. 276 (1663): 1823–1827. doi:10.1098/rspb.2008.1846. ISSN 0962-8452. PMC 2674493. PMID 19324785.
- ^ Azbukina, Nadezhda; Zharikova, Anastasia; Ramensky, Vasily (1 October 2022). «Intragenic compensation through the lens of deep mutational scanning». Biophysical Reviews. 14 (5): 1161–1182. doi:10.1007/s12551-022-01005-w. ISSN 1867-2469. PMC 9636336. PMID 36345285.
- ^ a b Burch, Christina L; Chao, Lin (1 March 1999). «Evolution by Small Steps and Rugged Landscapes in the RNA Virus ϕ6». Genetics. 151 (3): 921–927. doi:10.1093/genetics/151.3.921. ISSN 1943-2631. PMC 1460516. PMID 10049911.
- ^ a b Rimmelzwaan, G. F.; Berkhoff, E. G. M.; Nieuwkoop, N. J.; Smith, D. J.; Fouchier, R. A. M.; Osterhaus, A. D. M. E.YR 2005 (2005). «Full restoration of viral fitness by multiple compensatory co-mutations in the nucleoprotein of influenza A virus cytotoxic T-lymphocyte escape mutants». Journal of General Virology. 86 (6): 1801–1805. doi:10.1099/vir.0.80867-0. ISSN 1465-2099. PMID 15914859.
- ^ Kimura, Motoo (1 July 1985). «The role of compensatory neutral mutations in molecular evolution». Journal of Genetics. 64 (1): 7–19. doi:10.1007/BF02923549. ISSN 0973-7731. S2CID 129866.
External links[edit]
![]()
Wikimedia Commons has media related to Mutations.
- Jones S, Woolfson A, Partridge L (6 December 2007). «Genetic Mutation». In Our Time. BBC Radio 4. Retrieved 18 October 2015.
- Liou S (5 February 2011). «All About Mutations». HOPES. Huntington’s Disease Outreach Project for Education at Stanford. Retrieved 18 October 2015.
- «Locus Specific Mutation Databases». Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015.
- «Welcome to the Mutalyzer website». Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015. – The Mutalyzer website.