
Патологическая
физиология Д/З №2
Шуралёва Наталия
СВ – 21
-
ТЕОРИИ
СТАРЕНИЯ
Гипотезы
износа.
Наиболее
примитивные механистические гипотезы
рассматривали старение как простое
изнашивание клеток и тканей. Известность
получила одна из первых общебиологических
теорий, предложенная Н. Рубнером (1908).
Автор исходил из существования обратной
зависимости между интенсивностью
обмена, энергией и продолжительностью
жизни: «энергетическая теория старения».
Согласно расчетам Рубнера, количество
энергии на 1 кг массы тела, которое может
быть израсходовано за всю взрослую
жизнь, постоянно у всех животных, и
только человек имеет энергетический
фонд в 3—4 раза больший, чем другие
животные. Впоследствии это рассуждение
не подтвердилось для многих видов.
Неверным с точки зрения геронтологии
был и вытекающий отсюда вывод, что для
продления своей жизни человек должен
проявлять минимальную активность. На
самом деле ситуация противоположная,
и пассивный образ жизни сокращает ее
срок.
Гено-регуляторная
гипотеза.
Согласно
этой концепции первичные изменения
происходят в регуляторных генах —
наиболее активных и наименее защищенных
структурах ДНК. Предполагается, что эти
гены могут определять темп и
последовательность включения и выключения
тех генов (структурных), от которых
зависят возрастные изменения в структуре
и функции клеток. Прямых доказательств
возрастных изменений ДНК немного. В
последнее время высказывалось
предположение о связи старения с
участками ДНК, некоторые из которых
сокращаются в размерах при старении.
Сообщалось и об открытии особого
хромосомного фермента, препятствующего
старению ДНК и способного омолаживать
клетки человека (В. Райт и сотрудники).
Нейро-эндокринные
и иммунные гипотезы.
Нейроэндокринная
система является основным регулятором
жизненных функций организма. Поэтому
с самого начала в геронтологии активно
разрабатывались гипотезы, связывающие
ведущие механизмы старения на уровне
организма с первичными сдвигами в
нейро-эндокринной системе, которые
могут привести к вторичным изменениям
в тканях. При этом, более ранним
представлениям о первичном значении
изменений деятельности той или иной
конкретной железы (гипофиза, щитовидной
или, особенно, половых желез и т. д.)
приходят на смену взгляды, согласно
которым при старении изменяется функция
не одной какой-либо железы, а вся
нейро-эндокринная ситуация организма.
Довольно
широкую известность получили гипотезы,
связывающие старение с первичными
изменениями в гипоталамусе.
Согласно
гипотезе «гипоталамических часов»
(Дильман, 1968, 1976), старость рассматривается
как нарушение внутренней среды организма,
связанное с нарастанием активности
гипоталамуса. В итоге в пожилом возрасте
резко увеличивается секреция
гипоталамических гормонов (либеринов)
и ряда гормонов гипофиза (гонадотропинов,
соматотропина), а также инсулина. Но
наряду со стимуляцией одних структур
гипоталамуса, другие при старении
снижают свою активность, что приводит
к «разрегулированию» многих сторон
обмена и функции организма.
Молекулярно-генетические
гипотезы.
Наибольшее
внимание обычно привлекают
молекулярно-генетические гипотезы,
объясняющие процесс старения первичными
изменениями генетического аппарата
клетки. Большую их часть можно подразделить
на два основных варианта. В первом
случае, возрастные изменения генетического
аппарата клеток рассматриваются как
наследственно запрограммированные, во
втором — как случайные. Таким образом,
старение может являться запрограммированным
закономерным процессом, логическим
следствием роста и созревания, либо
результатом накопления случайных ошибок
в системе хранения и передачи генетической
информации.
Если
придерживаться первого мнения, то
старение, по сути, становится, продолжением
развития, в течение которого, в
определенной, закрепленной в эволюции
последовательности включаются и
выключаются различные участки генома.
Тогда при «растягивании» программы
развития замедляется работа «биологических
часов», задающих темп программе старения.
Например, в опытах с ограничением питания
в молодом возрасте (животные с «продленной
жизнью») происходит замедление роста,
а следовательно, и старения, хотя механизм
далеко не так прост. Предполагается,
что замедление роста и отодвигание
полового созревания и достижения
окончательных размеров тела приводит
к увеличению продолжительности жизни.
То есть, старение, как и другие этапы
онтогенеза, контролируется генами.
Гипотеза «накопления
ошибок»
Была
впервые предложена Л. Оргелем (1963). Она
основывается на предположении, что
основной причиной старения является
накопление с возрастом генетических
повреждений в результате мутаций,
которые могут быть как случайными
(спонтанными), так и вызванными различными
повреждающими факторами (ионизирующая
радиация, стрессы, ультрафиолетовые
лучи, вирусы, накопление в организме
побочных продуктов химических реакций
и другие). Гены, таким образом, могут
просто терять способность правильно
регулировать те или иные активности в
связи с накоплением повреждений ДНК.
В
то же время существует специальная
система репарации, обеспечивающая
относительную прочность структуры ДНК
и надежность в системе передачи
наследственной информации. В опытах на
нескольких видах животных показана
связь между активностью систем репарации
ДНК и продолжительностью жизни.
Предполагается ее возрастное ослабление
при старении. Роль репарации отчетливо
выступает во многих случаях преждевременного
старения и резкого укорочения длительности
жизни. Это относится, прежде всего, к
наследственным болезням репарации
(прогерии, синдром Тернера, некоторые
формы болезни Дауна и другие). В то же
время имеются новые данные о многочисленных
репарациях ДНК, которые используются
как аргумент против гипотез ошибок. В
статье под названием «Наука отрицает
старость» французский исследователь
Р. Россьон (1995) полагает, что в свете этих
фактов теория накопления ошибок в
нуклеотидных последовательностях.
требует пересмотра. Все же репарация,
видимо, не приводит к 100% исправлению
повреждений.
Многие
геронтологи считают, что старение —
результат накопления таких неисправленных
ошибок. По словам Хейфлика, «потеря
точной или надежной (контролирующей)
информации происходит из-за накопления
случайных воздействий, повреждающих
жизненно важные молекулы ДНК, РНК и
белков. Когда достигается пороговая
величина такого рода „поражений»,
„повреждений», „погрешностей»
или „ошибок», нормальные биологические
процессы прекращаются и возрастные
изменения становятся очевидными.
Истинная природа ущерба, наносимого
жизненно важным молекулам, пока
неизвестна, но известен сам факт его
проявления».
Некоторые
геронтологи, и среди них Ф. Маррот Сайнекс
из Медицинской школы Бостонского
университета, полагают, что ключевым
моментом в старении являются ошибки в
ДНК. Необратимые изменения в химической
структуре длинных, образующих ДНК
цепочек атомов получили название
мутаций. По Сайнексу, мутации—это
изменения в информации, зашифрованной
в структуре ДНК, которая контролирует
функционирование клетки. Мутации могут
возникать в результате неисправленных
ошибок при образовании повой ДНК, в
результате ошибок в процессе восстановления
или из-за повреждения ДНК загрязняющими
химическими веществами. Мутации в ДНК
клетки могут привести к тому, что клетка
начнет синтезировать измененную РНК,
а это в свою очередь приведет к синтезу
измененных белков — ферментов.
Видоизмененный фермент может работать
хуже нормального, а то и вовсе не работать.
В итоге реакции обмена веществ, в которых
участвует такой дефектный фермент,
могут прекратиться, и клетка перестает
выполнять свои функции или даже погибнет.
Теория
старения в результате накопления мутаций
впервые была выдвинута в 1954 г. физиком
Лео Сцилардом который пришел к этому
выводу, наблюдая за действием радиации
на людей и животных, сокращавшим их
жизнь. Радиация вызывает множественные
мутации ДНК, а также ускоряет появление
таких признаков старения, как седина
или раковые опухоли. Из этого Сцилард
сделал вывод, что именно мутации являются
причиной старения людей и животных. И
хотя он не сумел объяснить, каким образом
мутации возникают у людей и животных,
не подвергавшихся облучению, по его
мнению, они, возможно, есть не что иное,
как результат естественных повреждений
клеток.
Некоторые
современные геронтологи, в частности
д-р Говард Кёртис из Брукхвейнской
национальной лаборатории в Нью-Порке,
разделяют точку зрения Сциларда и также
считают, что старение вызывается
накоплением в течение жизни неисправленных
мутаций, разрушающих функциональные
потенции клетки. Кёртис полагает, что
старение, вызванное мутациями, можно
предотвратить или по крайней мере
замедлить, исправляя посредством генной
инженерии те процессы 11 клетках тела,
которые обусловливают репарацию (ремонт)
ДИК.
По
мнению некоторых ученых, обусловленное
мутациями ДНК старение не так серьезно,
как старение, вызванное неисправимыми
повреждениям и РНК, белков и ферментов
Д-р Лесли Оргел из Института Солка в
Ла-Хойе (Калифорния) предположил, что
ошибки в синтезе РНК и белков приводят
к старению клеток в результате, как он
это назвал, «катастрофы ошибок». Каждая
молекула РНК, считанная с ДНК, ответственна
за синтез множества копий определенного
фермента; РНК служит «матрицей», с
которой делается множество идентичных
копий молекулы белка. Следовательно,
при дефектной РНК каждая белковая
молекула, сходящая с «конвейера» будет
так же дефектна и не сможет эффективно
участвовать в реакциях обмена веществ.
Кроме того, некоторые ферменты участвуют
в производстве белков на базе «матричной»
РНК, а другие осуществляют синтез РНК
на матрице ДНК. Значит, если ошибка
вкралась в структуру РНК или белка, она
будет производить все более ущербные
«матрицы», что приведет к кумулятивному
эффекту — лавинообразному накоплению
ошибок и к последней катастрофе —
смерти.
Ученые
обнаружили, что действие ферментов из
культуры старых клеток ненормально: 25
% таких ферментов дефектны, что служит
подтверждением теории «катастрофы
ошибок» Оргела. И хотя это еще не
окончательное доказательство, можно
надеяться, что попытки предотвратить
старение, вызванное накоплением ошибок,
окажутся успешными. Возможно, понадобится
устранять не первичную ошибку на
молекулярном уровне, а лишь ее последствия.
Один из способов замедления аккумуляции
ошибок, который предлагает Алекс Комфорт,
заключается в некотором замедлении
скорости процессов обмена веществ и
клетках, что уменьшает вероятность
возникновения ошибки. Этого можно
добиться путем понижения температуры
тела. Как подтвердили опыты, жизнь
животных низших животных — рыб и черепах
— действительно от этого удлиняется.
6
Любой биологический организм со временем стареет, и человек, конечно же, не исключение. С возрастом каждый начинает замечать морщинки на лбу или снижение уровня энергии. Но что именно вызывает угасание человеческого тела? Перевели статью в журнале Business Insider, где представлены девять биологических процессов, совокупность действия которых мы и называем старением.
Чтобы выяснить, что же происходит внутри нашего тела и вызывает возрастные изменения и ухудшения, в 2013 году журнал Cell собрал группу исследователей. Команда, состоящая из ученых, которые изучали различные аспекты старения, провела масштабный анализ всей существующей литературы по старению и написала обзор. Работа «Признаки старения» (так называлась их статья) суммировала все биологическое, что происходит в нашем организме по мере того, как мы стареем. Ученые также разбили эти признаки на девять «процессов» (или причин. — Прим. ред.) старения — посмотрим, что означает каждый из них и как они работают.
В ДНК появляются ошибки
Один из видов повреждений, возникающих вместе со старением, — ошибки, которые начинают появляться в нашей ДНК. Когда ДНК реплицируется, ее код может не всегда копироваться правильно, в итоге некоторые ее части могут быть написаны с ошибками, а целые разделы могут быть случайно вставлены или удалены. Причем эти ошибки не всегда улавливаются механизмами в нашем организме, которые восстанавливают ДНК.
Генетический код — это инструкция для клетки, поэтому по мере накопления ошибок они могут нанести ущерб
Если со временем инструкции становятся неясными или неправильными, это может привести к разрушению клетки и даже к тому, что она станет злокачественной. Ученые обнаружили, что многие клетки в старых тканях имеют много накопленных генетических повреждений. Если исследователи смогут выяснить, как усовершенствовать механизм восстановления ДНК, они смогут улучшить и, возможно, замедлить процесс старения.
Неправильная экспрессия генов
Определенные части ДНК считываются и преобразуются в физические черты. Группа белков в клетках контролирует, какие гены в конечном итоге экспрессируются, — процесс называется эпигенетической модерацией, и именно он гарантирует, что клетки вашей кожи будут отличаться от клеток мозга, даже если они используют один и тот же набор ДНК.
Однако по мере того как мы стареем, белки, связанные с ДНК, становятся более рыхлыми и менее точными, и гены начинают экспрессироваться, когда этого не должно происходить, или замалчиваются по ошибке. В итоге некоторые необходимые белки не производятся, а вредные ненужные — наоборот. Например, если непреднамеренное изменение приводит к подавлению гена, который помогает подавлять опухоли, клетки могут неконтролируемо перерасти в рак. Ученые обнаружили, что устранение этих типов ошибок в экспрессии генов может улучшить некоторые неврологические эффекты старения у мышей, например ухудшение памяти.
Теломеры могут укорачиваться
Теломеры — это защитные колпачки на концах каждой цепи ДНК. Некоторые ученые сравнивают их с пластиковыми кончиками шнурков, которые предохраняют их от износа. Некоторые исследования показывают, что каждый раз, когда клетки делятся, кончики хромосом становятся чуть короче. Когда теломеры теряются, хромосомы становятся нестабильными, и возникают всевозможные проблемы. Наиболее примечательно то, что хромосомы не могут правильно реплицироваться и в конечном итоге становятся фрагментированными или у них появляются лишние части, которых там быть не должно. Такие аномалии обычно убивают клетки или делают их опасными.
Ученые выяснили, как повысить уровень теломеразы — фермента, который может увеличить длину теломер — у мышей, и их исследование показало, что это может продлить продолжительность жизни грызунов. Когда ученые снижали уровень теломеразы у мышей, продолжительность их жизни становилась короче.

Источник: istockphoto.com
Белки становятся менее стабильными и точными в своих ролях
Белки в наших клетках производятся постоянно, и именно они контролируют почти все функции внутри клетки. Они перемещают нужные «материалы», передают сигналы, включают и выключают процессы и обеспечивают структурную поддержку клетки. Но белки необходимо регулярно перерабатывать, потому что со временем они теряют свою эффективность.
С возрастом же наши тела теряют способность устранять старые белки
Когда наши тела не могут перерабатывать неиспользуемые белки, они накапливаются и могут стать токсичными. Накопление белка является одним из основных признаков болезни Альцгеймера: белки, называемые бета-амилоидными агрегатами в головном мозге, приводят к потере нервных клеток.
Клетки не умирают, когда должны
Когда клетки подвергаются стрессу и повреждаются, они иногда перестают делиться и становятся устойчивыми к смерти — превращаются в то, что ученые называют зомби-клетками, которые могут инфицировать другие клетки поблизости и распространять воспаление по всему телу. Такие клетки также называются стареющими клетками.
Со временем и возрастом стареющие клетки накапливаются, и ученые обнаружили, что устранение стареющих клеток у пожилых мышей, по-видимому, обращает вспять некоторые эффекты старения. Точно так же, когда стареющие клетки вводили молодым мышам, они оказывали изнурительное и воспалительное действие и наносили ущерб общему здоровью. В настоящее время разрабатывается несколько препаратов, называемых сенолитиками, которые должны уменьшать количество стареющих клеток у пожилых людей для лечения возрастных заболеваний.
Неисправности в механизме производства энергии
Энергию в клетках, превращая кислород и пищу в энергию, производят митохондрии, и по мере старения организма и его клеток эти миниатюрные электростанции становятся все более неэффективными и нефункциональными. Если же они не функционируют должным образом, они могут производить измененную форму кислорода, которая может вызвать повреждение ДНК и белков.
В исследовании, опубликованном в журнале Nature, ученые смогли обратить вспять появление морщин у мышей, восстановив функцию их митохондрий.
Обмен веществ может нарушиться
Клетки должны адаптироваться к доступному количеству питательных веществ, поэтому, если существует дисбаланс способности клетки воспринимать или обрабатывать питательные вещества, появляются проблемы.
С возрастом клетки становятся менее точными при определении количества глюкозы или жира в организме, поэтому некоторые виды жиров и сахаров не обрабатываются должным образом. То есть в стареющих клетках накапливается чрезмерное их количество не потому, что пожилые люди потребляют много жира, а потому, что клетки не переваривают его должным образом. В итоге это может повлиять на путь передачи инсулина и IGF-1, которые играют большую роль в возникновении диабета.
Именно поэтому возрастной диабет — это довольно распространенное явление: организм пожилых людей больше не может должным образом усваивать все, что они едят.

Источник: unsplash.com
Ткани перестают «чиниться» и обновляться
Почти все ткани в той или иной степени обновляются, но скорость такого обновления с возрастом замедляется, что является одной из причин накопления повреждений. Стволовые клетки — это клетки, которые могут превращаться в разные типы клеток нашего тела. Во многих тканях они действуют как внутренняя система восстановления, восполняя поврежденные или мертвые клетки. С возрастом стволовые клетки истощаются и становятся менее активными, а это означает, что они больше не могут делиться так быстро. Истощение стволовых клеток означает, что ткани, которые должны обновляться, на самом деле больше не делают этого.
Клетки перестают «общаться» друг с другом
Чтобы все в организме работало, клетки должны постоянно «общаться» друг с другом — для этого они посылают сигналы через кровь и иммунную систему. Но по мере того как наши тела стареют, клетки начинают делать это все хуже. Так, некоторые из них становятся менее чувствительными, что может превратить их в стареющие клетки, вызывающие воспаление. При этом само это воспаление еще больше блокирует связь со здоровыми функционирующими клетками.
Когда клетки не могут «общаться», иммунная система не может эффективно избавляться от патогенов и стареющих клеток
Старение также изменяет уровень межклеточной коммуникации в эндокринной и нейроэндокринной системах. То есть сообщения, отправляемые через молекулы гормонов, циркулирующих в этих системах, таких как инсулин, обычно теряются.

Ученые пока не понимают связи между этими девятью согласованными признаками старения, но, по словам *Мануэля Серрано *, одного из авторов статьи, сейчас многие ученые занимаются подобными исследованиями.
Когда ученые в достаточной мере узнают о процессах, лежащих в основе старения, они смогут создавать более эффективные методы лечения, которые могут влиять на то, как мы стареем, и лечить возрастные заболевания.
Ричард Миллер, директор Центра исследований старения Гленна при Мичиганском университете, говорит, что, когда дело доходит до управления старением, «вещи, которые действительно имеют значение, — это основные механизмы контроля в нашем теле, которые регулируют различные виды клеточных событий». По его мнению, настоящая проблема состоит в том, чтобы выяснить, что связывает воедино все процессы, вызывающие разрушение нашего тела.

Старение — основной фактор риска развития многих распространенных заболеваний. Болезни преждевременного старения человека являются перспективными модельными системами для определения и описания клеточных механизмов, лежащих в основе физиологического старения. Также их изучение позволяет лучше понять причины, пусковые факторы и потенциальные стратегии лечения распространенных болезней старости, в том числе неврологических расстройств, диабета, онкологических и сердечно-сосудистых заболеваний. Используя в качестве основной парадигмы преждевременного старения редкую патологию — синдром Хатчинсона-Гилфорда, авторы обсудят общие механизмы преждевременного старения и болезней старости, включая генетические и эпигенетические дефекты, дефекты в метаболических путях; митохондриальный и белковый гомеостаз; клеточный цикл и способность стволовых клеток к регенерации.
Старение — это процесс постепенного функционального истощения на клеточном и организменном уровнях. Болезни, связанные со старением (ageing-associated diseases, AADs), к которым относится большинство патологий сердечно-сосудистой системы, хроническая обструктивная болезнь легких (ХОБЛ), инсульт, болезнь Альцгеймера, хроническая болезнь почек (ХБП) и онкологические заболевания, являются причиной приблизительно половины всех человеческих смертей (см. «Хронические заболевания, связанные со старением») [1,2]. Хотя старение — основной фактор риска развития этих заболеваний, понимание того, как именно оно участвует в их возникновении и развитии, находится в зачаточном состоянии.
Роль старения в развитии болезней человека в основном изучается на животных моделях заболеваний, путем препятствования началу развития и прогрессированию дефектов в тканях, преимущественно вовлеченных в патогенез различных связанных со старением болезней (см. Вставка 1). Несмотря на то, что животные модели являются удобной заменой для изучения основ старения, которые могут быть одинаковы для разных видов, они не идеальны для освещения эффектов старения на человеческие болезни, в силу слишком низкой встречаемости AADs у лабораторных животных в сравнении с людьми [3–5].
Ключевая причина ААDs — снижение функции клеток и тканей, связанное со старением [6,7]. Клеточное старение характеризуется повышением нестабильности генома, изменением метаболизма и потерей регенеративного потенциала. Рассмотрение старения и износа клеток в качестве движущей силы болезней, связанных со старением, позволяет объяснить наблюдаемое при AADs поражение не только ткани, непосредственно вовлеченной в патологический процесс, но и одновременное снижение функции других тканей [6,7]. Функциональный износ тканей многих органов, часто упускаемый из виду при AADs, важен для диагностики и изучения патологии: например, сила рукопожатия и переломы бедра являются индикаторами ХПН и болезни Паркинсона, соответственно [8,9]. Эти наблюдения показывают, что клеточное старение является универсальным принципом, лежащим в основе заболеваний, связанных со старением.
Вставка 1 | Хронические заболевания, связанные со старением
Болезнь Альцгеймера
Хроническое нейродегенеративное заболевание, характеризующееся деменцией, дезориентацией, перепадами настроения, потерей аппетита, сумбурностью речи и неспособностью к координации движений; эта болезнь также ассоциирована с повышенным риском остеопороза и мышечной атрофии. В большинстве случаев семейные формы болезни Альцгеймера обусловлены мутациями в белке-предшественнике β-амилоида (APP) и в пресенилинах 1 и 2, которые усиливают образование β-амилоида — продукта расщепления APP (обычно откладывающегося в сенильных бляшках). Кроме этого, нейрофибриллярные клубки, состоящие в основном из гиперфосфорилированного тау-белка, являются ключевым признаком болезни Альцгеймера.Атеросклероз
Заболевание сосудов, характеризующееся тем, что артерии становятся менее эластичными и кальцифицируются, вследствие образования холестериновых бляшек, которые препятствуют току крови. Нестабильные бляшки имеют меньшее число гладких миоцитов и более склонны к разрыву, что может приводить к инфаркту или инсульту.Рак
Группа заболеваний, подразумевающих под собой аномальный рост клеток, что проявляется либо в инвазивной форме (злокачественной), либо неинвазивной (доброкачественной), как результат накопления генетических мутаций, ингибирующих активность генов-супрессоров опухолей, или активацией/оверэкспрессией онкогенов.Хроническая болезнь почек
Хроническое состояние, определяемое как постепенное снижение функций почек, способное приводить к повышению кровяного давления, анемии, уменьшению костной массы и повреждению нейронов.Хроническая обструктивная болезнь легких
Группа легочных патологий, при которых уменьшается воздушный поток и затрудняется дыхание, что обусловлено в основном повышением продукции слизи и воспалением (бронхит) либо же разрушением альвеол и расширением воздушных пространств легких (эмфизема). Идиопатический легочный фиброз характеризуется утолщением и рубцеванием легочной ткани, приводящих к ухудшению газообмена между легкими и кровью. Пациенты с ХОБЛ имеют повышенный риск развития болезни Паркинсона.Сердечная недостаточность
Хроническое состояние, характеризующееся сниженным сердечным выбросом, причиной которого является неспособность сердца адекватно сокращаться вследствие растяжения/истончения стенки желудочков, или же расслабляться из-за их гипертрофии / сниженной эластичности.Остеопороз
Уменьшение костной массы из-за дисбаланса между процессами остеогенеза и резорбции кости.Болезнь Паркинсона
Хроническое нейродегенеративное заболевание, при котором нарушается функция моторной системы, что приводит к тремору, ригидности, нарушениям походки и зачастую к деменции и депрессии. Причины наследственных форм болезни Паркинсона включают в себя мутации α-синуклеина, parkin, серин/треониновой богатой лейциновыми повторами протеинкиназы 2 (LRRK2), PTEN-индуцированной киназы 1 (PINK1), DJ1 и TP13A2. Одним из типичных признаков болезни Паркинсона является накопление α-синуклеина в виде телец Леви, которые способствуют гибели клеток в имеющем дофаминергическую иннервацию черном веществе.Саркопения
Потеря мышечных массы и силы в ходе старения. Пациенты с ХОБЛ, сердечной недостаточностью, раком или ХБП имеют повышенный риск саркопении.Сахарный диабет 2 типа
Метаболическое расстройства, при котором инсулин не может быть использован должным образом, что частично компенсируется повышением его образования панкреатическими β-клетками. В конце счете последние клетки перестают справляться. Пациенты с СД2 находятся в группе риска развития болезни Альцгеймера и ХБП.
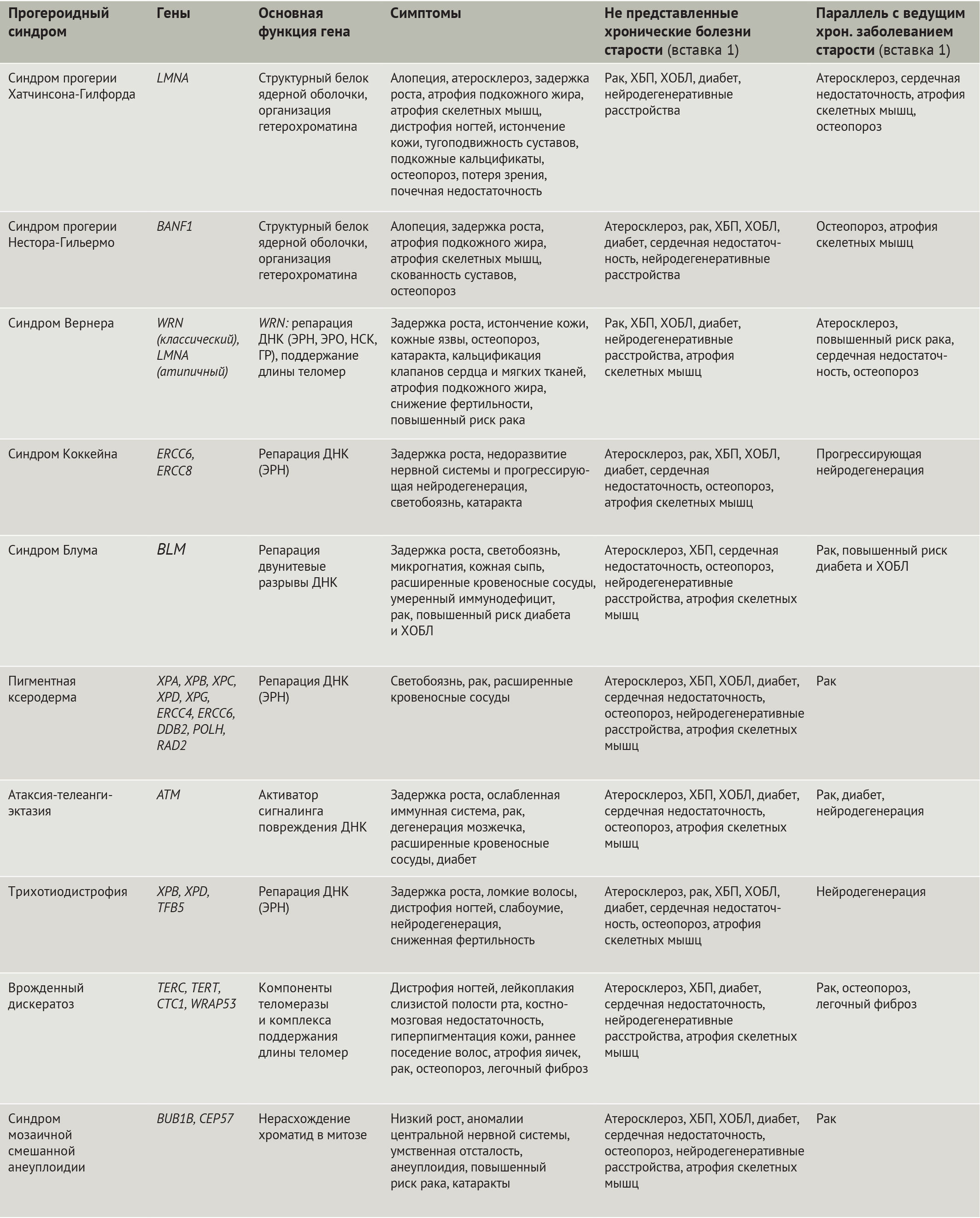
Значительно облегчило изучение старения человека открытие мутаций, лежащих в основе синдромов преждевременного старения (прогероидных синдромов, — прим. ред.) (табл. 1). Наиболее яркими из них являются: синдром преждевременного старения Хатчинсона-Гилфорда (СПХГ) [10–12] и атипичный синдром Вернера, обусловленные дефектами белков ядерной оболочки (равно как и классическая форма синдрома Вернера), синдромы Коккейна, Блума, пигментная ксеродерма, атаксия-телеангиэктазия, трихотиодистрофия, врожденный дискератоз (ВДК), синдром мозаичной смешанной анеуплоидии, причиной которых являются дефекты в системе репарации ДНК, в т.ч. и в обеспечивающих ее белках (табл. 1). Клеточные дефекты, наблюдаемые при этих и других болезнях преждевременного старения, включая нестабильность генома и протеома, изменение метаболизма и потерю регенеративного потенциала, совпадают с таковыми при физиологическом старении организма. Кроме того, существуют поразительные сходства между дефектами организма при некоторых прогероидных синдромах и AADs. Тем не менее, т.к. синдромы преждевременного старения отображают лишь некоторые аспекты физиологического старения человека, неудивительно, что прогероидные патологии имеют общее лишь с некоторыми из AADs (табл. 1). К примеру, при СПХГ наблюдаются значительные нарушения сердечно-сосудистой системы и остеопороз, в то время как болезни преждевременного старения, связанные с дефектами белков репарации ДНК, характеризуются предрасположенностью к онкологическим и нейродегенеративным заболеваниям. Наличие таких параллелей предполагает общность этиологии у прогероидных синдромов и AADs.
Таблица 1 | Прогероидные синдромы
В данном обзоре авторы обсудят дефекты в клеточных и молекулярных механизмах, общих для болезней преждевременного старения и для физиологического старения, а также подробно осветят роли этих путей в развитии некоторых болезней, ассоциированных со старением. В качестве парадигмы взаимосвязи между преждевременным и физиологическим старением и AADs авторы рассмотрят СПХГ, в силу того, что это — одна из наиболее полно изученных болезней преждевременного старения, демонстрирующая широкий спектр дефектов на клеточном, тканевом и организменном уровнях, общих с AADs.
СПХГ — редчайшая болезнь преждевременного старения с частотой встречаемости 1 случай на 4–8 млн новорожденных [13]. Клинические особенности СПХГ проявляются уже вскоре после рождения и включают: тотальную алопецию, атрофию подкожно-жировой клетчатки и скелетных мышц, ониходистрофию, тугоподвижность суставов, появление морщин и подкожных кальцификатов, нарушения структуры костей и потерю зрения (табл. 1) [13]. Болезнь неминуемо приводит к гибели в возрасте 15–17 лет, при этом основными причинами смерти являются атеросклероз-подобная прогрессирующая патология сердечно-сосудистой системы, инфаркт миокарда и инсульт [14]. СПХГ в основном обусловлен гетерозиготной сайленс-мутацией (G608G) в гене LMNA, приводящей к синтезу прогерина — структурного ядерного белка ламина А, аномальной формы [10,11,15].
Ламин А дикого типа претерпевает посттрансляционный процессинг: при отщеплении С-конца под действием гомолога CAAX-пренил-протеазы 1 (кодируемой геном ZMPSTE24, также известным у мышей как ZMPSTE24) образуется зрелый ламин А, молекулы которого организуются в промежуточные филаменты, далее встраивающиеся в ядерные ламину и матрикс (рис. 1) [16,17]. При СПХГ мутация 1824С>T (LMNAG608G) активирует криптический (скрытый, — прим. ред.) сайт сплайсинга, что приводит к удалению 50-аминокислотной последовательности, содержащей сайт узнавания ZMPSTE24. В результате этого прогерин накапливается на периферии ядра и вызывает нарушения механохимических свойств ламины, что можно понять по аномальной форме ядра, наблюдаемой в клетках у пациентов с СПХГ (рис. 1). Подобные клеточные дефекты обусловлены мутациями LMNA, вызывающими атипичные формы СПХГ (включая LMNAG608S и LMNAE145K) [16,18]. Нарушения механических свойств и механотрансдукции ядерной ламины, как считается, играют роль в патологии у пациентов с СПХГ, т. к. многие поражаемые ткани, такие как сосуды, кости и суставы, особенно подвержены механическим воздействиям [17,19].
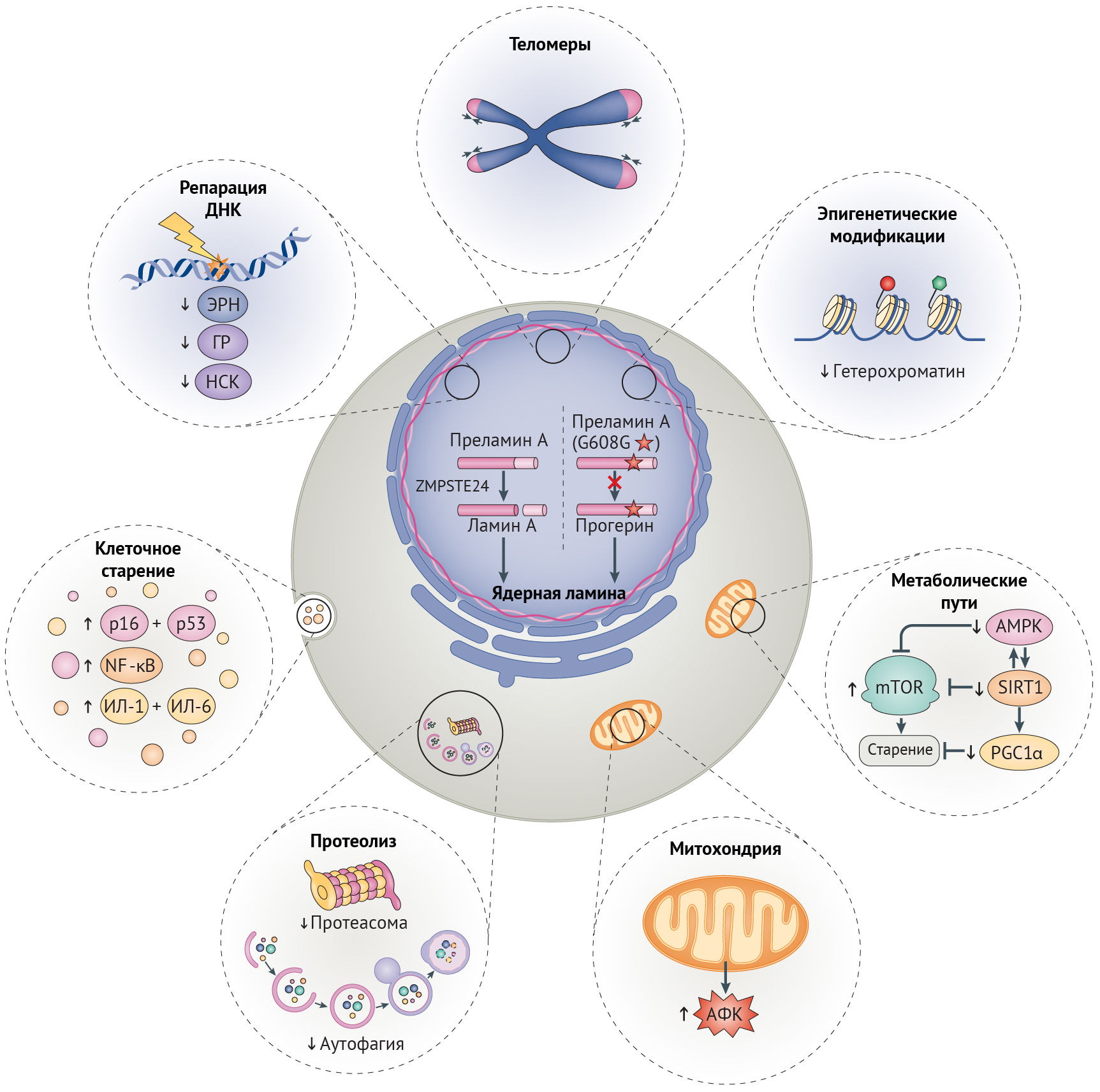
Рисунок 1 | Дефекты клеточного старения
Общая схема дефектов клеточного старения, лежащих в основе синдромов преждевременного старения, нормального старения и заболеваний старости. Эти дефекты старения связаны между собой и включают в себя снижение эффективности путей репарации ДНК, потерю геномной целостности, уменьшение доли гетерохроматина, сдвиги в метаболическом сигналинге, усиление образования митохондриями активных форм кислорода (АФК), недостаточность работы протеолитических путей, поддерживающих протеостаз и активацию путей клеточного старения. В результате посттрансляционного процессинга преламина А дикого типа цинковой металлопротеазой ZMPSTE24 образуется зрелый белок, ламин А, встраивающийся в ядерную ламину (на рисунке в левой половине ядра). При синдроме преждевременного старения Хатчинсона-Гилфорда мутация в гене LMNAG608G приводит к активации скрытого сайта сплайсинга, результатом чего является образование изоформы преламина А с недостатком 50-ти аминокислот, включая сайт сплайсинга, распознаваемый ZMPSTE24. Эта изоформа получила название прогерин (на рисунке правая половина ядра). Включение ее в ядерную ламину приводит к искажению формы ядра и в дальнейшем участвует в возникновении других дефектов старения, отображенных на рисунке (пути и механизмы с нарушением регуляции показаны тонкими стрелками). AМФК, АМФ-активируемая протеинкиназа; ГР, гомологичная рекомбинация; ИЛ-1, интерлейкин-1; НСК, негомологичное соединение концов; ЭРН, эксцизионная репарация нуклеотидов; NF-κB, ядерный фактор-κB; PGC1α, коактиватор 1α γ-рецептора, активируемого пролифераторами пероксисом; SIRT1, сиртуин 1.
СПХГ отличается от других прогероидных синдромов своим ранним началом, выраженностью признаков старения и большим числом вовлеченных в патологический процесс тканей (табл. 1) [15,20]. В отличие от СПХГ, классическая форма синдрома Вернера (прогерии взрослых) проявляется на третьем десятилетии жизни остеопорозом и онкологическими заболеваниями.
Раннее проявление дефектов старения у больных СПХГ, вероятно, отчасти связано с доминантно-негативным действием прогерина, в то время как другие синдромы преждевременного старения зачастую ассоциированы с рецессивными мутациями, приводящими к утрате функции белков репарации ДНК, что может быть частично скомпенсировано альтернативными путями репарации [20]. Как следствие, у больных СПХГ наблюдается широкий спектр тканевых дефектов (от остеопороза до связанных со старением патологий скелетных мышц и сердечно-сосудистой системы). В свою очередь прогероидные синдромы, вызванные нарушением репарации ДНК, в своей массе ассоциированы с высокой предрасположенностью к онкозаболеваниям и, в некоторых случаях, с прогрессирующей нейродегенерацией (табл. 1) [20]. Любопытно, что по непонятным причинам у пациентов с СПХГ не повышен риск развития диабета и ХБП. Кроме того, у них не происходит и нейродегенерации; вероятно, причина этого — микроРНК-9 (miR-9), которая подавляет экспрессию ламина А и прогерина в нейронах. Также несмотря на значительные повреждения ДНК, у пациентов с СПХГ не развиваются опухоли в детском возрасте, что может быть связано с изменением структуры внеклеточного матрикса, препятствующим инвазии опухоль-инициирующих клеток [21,22], и с тем, что прогерин оказывает протективный эффект в отношении опухолевого роста [23].
Несмотря на то, что некоторые симптомы заболеваний, связанных с возрастом не наблюдаются при СПХГ (табл. 1), многие ключевые признаки клеточного старения, отмечаемые в тканях, поражаемых при AADs, встречаются и у пациентов с СПХГ (рис. 1) [24]. Это яркое проявление дефектов старения при СПХГ было объяснено негативным действием прогерина на ядерную ламину, являющейся основным каркасом клеточного ядра млекопитающих [25]. Установлено, что дефекты ламины участвуют в механизме возникновения большинства признаков, общих для старения и AADs (рис. 1), включая утрату генетической и эпигенетической целостности, укорочение теломер, нарушение гомеостаза белков (протеостаза), перепрограммирование метаболизма, митохондриальную дисфункцию, клеточное старение, нарушение поддержания функции стволовых клеток и их регенеративного потенциала [26]. Таким образом, экспрессия прогерина вовлечена в различные механизмы, в то время как при прогероидных синдромах Вернера, Блума и Коккейна наблюдается более ограниченный спектр дефектов, обусловленных в основном нарушением репарации ДНК [27–34]. Важность ядерной ламины в старении иллюстрируется еще одной болезнью преждевременного старения — синдромом прогерии Нестора-Гильермо (СПНГ), причиной которого служит гомозиготная мутация в гене белка, локализованном в ядерной ламине, — фактора барьера аутоинтеграции 1 (BANF1) [35]. Подобно СПХГ, при СПНГ наблюдаются тугоподвижность суставов, дистрофия подкожно-жировой клетчатки и скелетных мышц и отставание в росте (рис. 1) [35]. Причастность дисфункции ядерной ламины к различным признакам клеточного старения дает возможность рассматривать СПХГ как подходящую модель для обсуждения параллелей между преждевременным старением и AADs на клеточном и молекулярном уровнях.
СПХГ имеет особенное отношение к обычному старению, потому как у физиологически стареющих людей также вырабатывается небольшое количество прогерина — благодаря периодически происходящему самопроизвольному открытию криптического сайта сплайсинга, который активирован у пациентов с СПХГ [28, 36]. В силу доминантно-негативного действия прогерина, его небольшие количества, вероятно, участвуют в процессе нормального старения [36]. Эта точка зрения подтверждается высокой схожестью клеточных и организменных дефектов у пациентов с СПХГ и физиологически стареющих индивидов [20,26]. Вдобавок к этому, устранение экспрессии прогерина и ламина А на генетическом уровне увеличивает продолжительность жизни мышей [37], а мутация 1968G>A в гене LMNA, приводящая к более слабой активации скрытого сайта сплайсинга прогерина, своим следствием имеет менее яркую выраженность связанных со старением патологий [38].
Единственный, к тому же достоверно определенный, генетический дефект у пациентов с СПХГ, прямая связь между прогерином и многочисленными признаками клеточного старения, экспрессия патогенного белка в ходе физиологического старения и манифестация связанных со старением патологий во многих тканях — все это делает СПХГ привлекательной моделью для изучения роли механизмов старения в патогенезе AADs.
Человеческий геном постоянно подвергается влиянию факторов, повреждающих ДНК и угрожающих клеточному гомеостазу. Этому противодействуют механизмы, исправляющие повреждения и не дающие укорачиваться концам хромосом. Снижение их эффективности с возрастом и при AADs приводит к износу клетки [39–42].
Целостность генома поддерживают несколько механизмов репарации ДНК. Механизм эксцизионной репарации нуклеотидов (NER, ЭРН) исправляет повреждения на одной цепи ДНК [40–42], в то время как двунитевые разрывы (DSBs, ДНР), приводящие к крупным хромосомным перестройкам и угрожающие выживанию клетки, в первую очередь и с высокой точностью исправляются с помощью гомологичной рекомбинации, но могут репарироваться и методом негомологичного соединения концов (NHEJ, НСК), подверженному ошибкам. Эффективность этих механизмов репарации снижается в процессе старения (рис. 2) [39–42], результатом чего служит повышение с возрастом количества хромосомных аберраций и перманентной активации сигналинга повреждений ДНК [43].
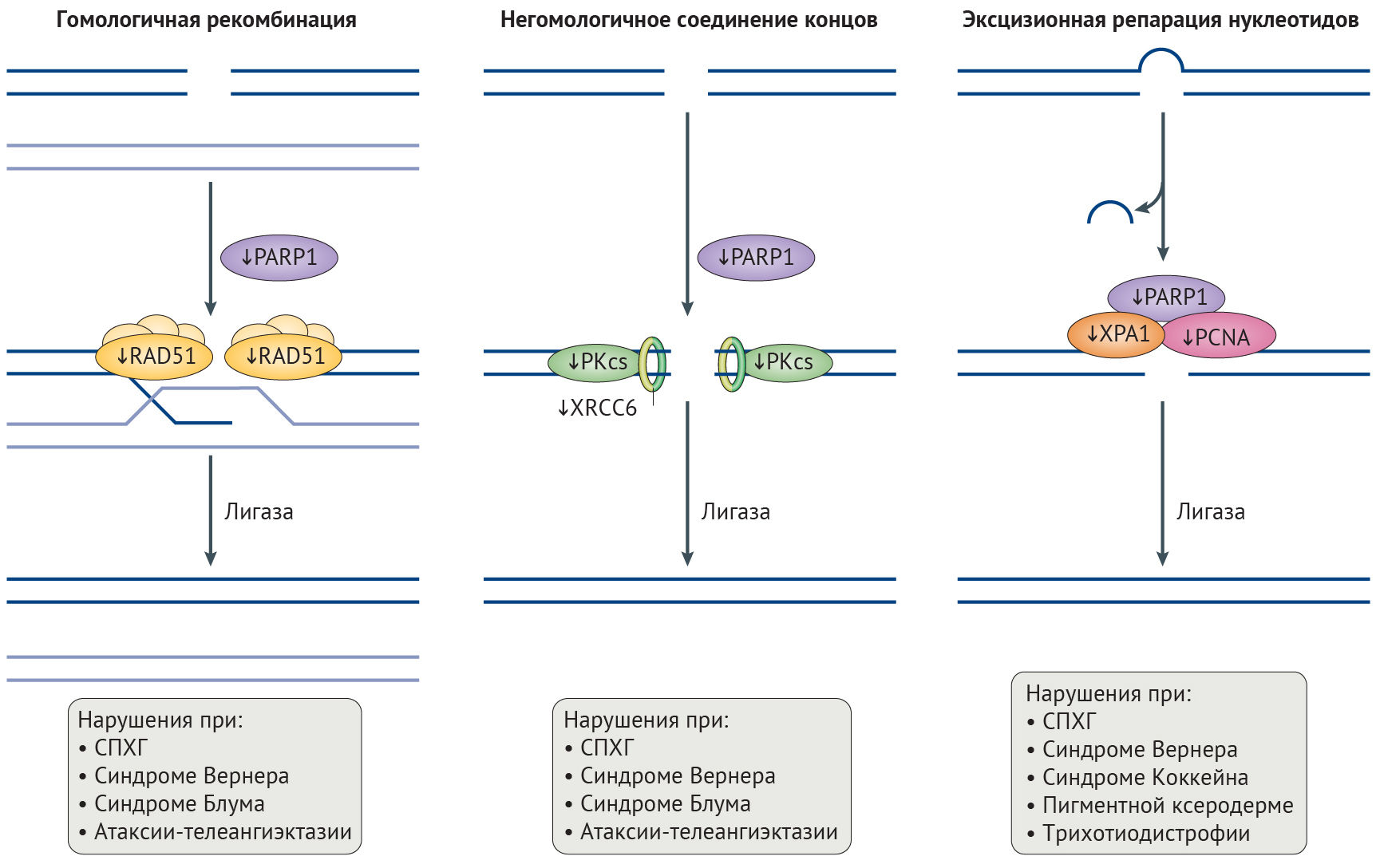
Рисунок 2 | Дефекты в репарации ДНК, связанные со старением
Общая схема путей репарации ДНК, дефектных при синдромах преждевременного старения, нормального старения и при болезнях старости. Нарушения в белках репарации, специфичные для синдрома прогерии Хатчинсона-Гилфорда (СПХГ), показаны тонкими стрелками. Двухцепочечные разрывы исправляются как путем гомологичной рекомбинации посредством RAD51, при которой сестринская хромосома используется как матрица, так и путем негомологичного соединения концов, с помощью перекрестно-комплементирующего белка репарации рентгеновских повреждений (XRCC6) и каталитической субъединицы ДНК-зависимой протеинкиназы (PKcs). Поли(АДФ-рибоза)-полимераза 1 (PARP1) способствует активации опосредованного ATM (мутантны при атаксии-телеангиэктазии) сигналинга повреждения ДНК. При эксцизионной репарации нуклеотидов объемные аддукты ДНК вырезаются, и образующиеся в результате одноцепочечные разрывы ДНК репарируются указанными белками. Конечный этап репарации для каждого из ее путей заключается в соединении цепей ДНК в точке разрыва при помощи лигазы.
PCNA, ядерный антиген пролифелирующих клеток.
Неполноценность репарации ДНК при СПХГ. Хроническая активация сигналинга повреждений ДНК при СПХГ приводит к повышению числа ДНР и неполноценному их исправлению [44]. Дефекты репликации, включая коллапс репликативной вилки, играют большую роль в появлении ДНР и особенно часто наблюдаются при СПХГ. Причиной этого служат уменьшение содержания одного из необходимых для репликации ДНК белков скользящего зажима — ядерного антигена пролиферирующих клеток (PCNA), а также нарушение взаимодействий между ним и ламином А и повышение активности протеолитической деградации фактора репликации C1 (рис. 2) [45–47]. Эффективность исправления ДНР, осуществляемого с помощью механизмов гомологичной рекомбинации и НСК, снижена в силу замедленного рекрутинга белков репарации (таких как TP53-связывающий белок 1 (TP53BP1) и RAD51), ингибирования белка репарации ДНК поли(АДФ-рибоза)-полимеразы 1 (PARP1), а также пониженного содержания участвующих в НСК белков — DNA-PKcs и перекрестно-комплементирующего белка репарации рентгеновских повреждений (XRCC6) [30,48,49]. Более того, некорректное связывание белка репарации XPA в участках ДНР также препятствует репарации и уменьшает эффективность ЭРН (рис. 2) [45].
Неполноценность репарации ДНК при других прогероидных синдромах. О роли ЭРН и репарации ДНР в процессе старения и патогенезе AADs убедительно говорят прогероидные фенотипы при синдроме Коккейна, пигментной ксеродерме, трихотиодистрофии (вызываемых дефектами белков ЭРН) и атаксии-телеангиэктазии, синдромах Вернера и Блума (вызываемых нарушением репарации ДНР) (рис. 2; табл. 1) [20]. Высокий процент патологий нервной системы среди пациентов с некоторыми из этих прогероидных синдромов говорит о том, что жизнедеятельность постмитотических нейронов существенно зависит от эффективности репарации ДНК, что, вероятно, связано с накоплением повреждений ДНК в течение жизни организма, причиной чего являются высокая транскрипционная активность и воздействие оксидативного стресса [50]. Удивительно, но эффективность системы репарации ДНК в головном мозге низка, и становится еще ниже при нейродегенеративных заболеваниях [50]. Это наблюдение согласуется с повышенной встречаемостью анеуплоидных нейронов при болезни Альцгеймера [50,51], вероятно, связанной с дефектами НСК, что хорошо соотносится с данными о прямом ингибировании ЭРН мутантным белком parkin при наследственной форме болезни Паркинсона (см. Вставка 1) [52]. Более того, при обеих данных патологиях фибробласты и лимфобласты обладают повышенной чувствительностью к вызванным рентгеновским излучением повреждениям ДНК [50,53]. Увеличенный уровень повреждения ДНК может быть причиной как функциональных нарушений в нейронах, так и уменьшения числа нервных клеток вследствие индукции апоптоза [50].
Неполноценность репарации ДНК при AADs. Повышение уровня повреждений ДНК наблюдается и в некоторых специфических типах клеток, поражаемых при AADs. В гладких миоцитах сосудов (ГМС), повреждаемых при атеросклерозе, уровень повреждений ДНК при воздействии активных форм кислорода (АФК) выше, вследствие возрастания механического стресса, накопления липопротеинов и воспалительного ответа. Последующее снижение пролиферации и апоптоз ГМС может привести к разрыву атеросклеротической бляшки [54]. ГМС очень быстро «стареют» у пациентов с СПХГ, что сочетается с дефектами гомологичной рекомбинации [48]. Подобно этому, лимфоциты у больных диабетом второго типа (СД2) обладают сниженной способностью исправлять повреждения ДНК, вызванные АФК, а β-клетки поджелудочной железы страдают из-за недостаточности системы НСК [55,56]. Более того, дифференцировка образующих кость остеобластов нарушается при повреждениях ДНК, что является причиной хрупкости костей у мышиных моделей прогерии [57]. Согласно этим данным логично предположить, что дефектное НСК может приводить к остеопении, менее тяжелой старческой форме остеопороза [58].
Пониженная эффективность репарации ДНК ведет к накоплению геномных мутаций в ходе старения, что способствует опухолеобразованию благодаря активации онкогенов или инактивации генов-супрессоров опухоли. Действительность этого механизма подтверждается высокой встречаемостью опухолей при большинстве прогероидных синдромов с нарушениями репарации ДНК (табл. 1) [20].
Примеры выше наводят на мысль, что повреждения ДНК являются ключевым фактором развития AADs, но главный вопрос остается открытым: стохастически ли влияет репарация ДНК на целостность генома, или же она затрагивает специфические участки последнего [59].
Теломеры — повторяющиеся последовательности на концах хромосом, прикрытые шелтериновым комплексом, включающим в себя факторы, которые связываются с повторами теломер 1 и 2 (TERF1 и TERF2), и препятствующим распознаванию концов хромосом в качестве ДНР [60]. Теломеры укорачиваются в ходе деления клетки (процесс, известный как истощение теломер) и при достижении критического размера могут активировать сигнальные каскады повреждения ДНК и запускать старение клетки. Теломераза — комплекс, состоящий из обратной транскриптазы (TERT) и теломеразного РНК-компонента (TERC), который удлиняет теломеры при каждом клеточном цикле. Она активно экспрессируется в стволовых клетках эмбриона, но в то же время не обнаруживается в большинстве других человеческих клеток [61].
У пациентов с СПХГ определяется повышенная активация сигналинга о повреждении ДНК; их теломеры короче [62, 63], а число хромосомных аберраций — выше из-за дефектной гомологичной рекомбинации [64]. Подобно этому у прогероидных мышиных моделей с нокаутом гена Terc наблюдается соединение хромосом конец в конец [20]. Прогерин в свою очередь может усиливать повреждения теломер, нарушая ламин А и TERF2-зависимые системы стабилизации концов теломер, что и происходит у пациентов с атипичным синдромом Вернера [65–68].
Повреждения теломер могут запускать синтез прогерина в клетках дикого типа, а потеря функций теломеразного комплекса лежит в основе прогероидного синдрома ВДК, исходя из чего можно предположить, что укорочение теломер имеет определяющую роль в развитии AADs (табл. 1) [20,69]. Генетическая абляция РНК Terc у мышей приводит к укорочению теломер до критической длины спустя несколько поколений, следствием чего является развитие гипертрофии кардиомиоцитов, снижение функции левого желудочка и повышение систолического артериального давления, характерные для физиологического старения [61]. Кроме того, у мышей с нокаутом Terf2 наблюдается усиленный апоптоз кардиомиоцитов [60]. Роль укорочения теломер в патогенезе заболеваний сердечно-сосудистой системы подтверждается и атеросклеротическим фенотипом у мышей с нокаутом Terc, а также наблюдением, что и у человека эндотелиоциты и ГМС в регионах, подвергающихся повышенному гемодинамическому стрессу, и в атеросклеротических областях имеют более короткие теломеры [60]. Повышенная активность теломеразы может быть как полезна, т. к. повышается срок жизни эндотелиоцитов, так и вредна, потому что способствует пролиферации лейкоцитов и образованию в интиме новых ГМС, тем самым обостряя течение атеросклероза [60].
Истощение теломер также является причиной остеопороза у пациентов с ВДК. Теломеры в хондроцитах и лейкоцитах периферической крови у пожилых больных остеопорозом обладают меньшей длиной, а восстановление активности теломеразы в остеобластах усиливает образование костной ткани [70]. Мутации, приводящие к потере функции TERT и истощению теломер, обуславливают развитие и старческих эмфиземы и фиброза легких [71]. Исследования на мышиных моделях дают возможность предположить, что укорочение теломер ускоряет развитие старческой эмфиземы благодаря уменьшению способности легких противостоять стрессу, индуцированному токсинами [72,73]. Было обнаружено, что при ХБП истощение теломер снижает жизнеспособность клеток и усиливает повреждение почек в ходе ишемии-реперфузии [71]. Вполне возможно, это действительно и для β-клеток поджелудочной железы, выживаемость и функциональность которых снижена при высоком уровне сахара в крови у мышей с нокаутом Tert [71].
Хотя экспрессия TERT в фибробластах у больных СПХГ и может нивелировать некоторые клеточные дефекты, связанные со старением, наблюдение, что у экспрессирующих TERT и прогерин человеческих фибробластов тем не менее проявляются некоторые дефекты старения, позволяет говорить о наличии и других механизмов, задействованных в клеточном старении — регулирующих целостность теломер и других участков генома или запускающих старение [32,74].
Регуляция структуры хроматина достигается с помощью метилирования ДНК и посттрансляционных модификаций гистонов, что влияет на целостность генома, экспрессию генов и в конечном счете на здоровье клетки и развитие заболеваний [26,75].
В клетках стареющих лиц наблюдается снижение содержания гистонов в целом и постепенное снижение активности триметилирования лизина в 9 и 27 положениях гистона H3 (H3K9me3 и H3K27me3) — репрессивных меток, способствующих компактизации хроматина (рис. 3) [26]. Утрата гетерохроматина усиливается при характерном для старения ингибировании α-гомолога белка гетерохроматина 1 (HP1α, также известного как CBX5), комплекса ремоделирования и деацетилирования нуклеосом (NuRD) и белков группы Polycomb (PCG), каждый из которых является эпигенетическим сайленсером [20,32,76]. Уменьшение доли гетерохроматина, белков-участников комплекса NuRD и HP1α было отмечено и в клетках пациентов с СПХГ [28, 77]. Потеря комплекса NuRD и сниженная экспрессия H3K27-специфичной метилтрансферазы EZH2 в этих клетках приводит к потере репрессивной метки H3K27me3 гетерохроматина (рис. 2) [32,77]. Любопытно, что нокдаун H3K27-деметилазы UTX1 у Caenorhabditis elegans (один из видов свободноживущих нематод, — прим. ред.) увеличивает продолжительность жизни последних на 30 % [78]. Сниженная активность EZH2, компонента ингибиторного комплекса Polycomb 2 (PRC2), играет роль в развитии СД2, поскольку кондиционная делеция EZH2 в панкреатических β-клетках молодых мышей индуцирует клеточное старение, что приводит к уменьшению количества и пролиферации этих клеток и тем самым обуславливает диабетический фенотип [79].
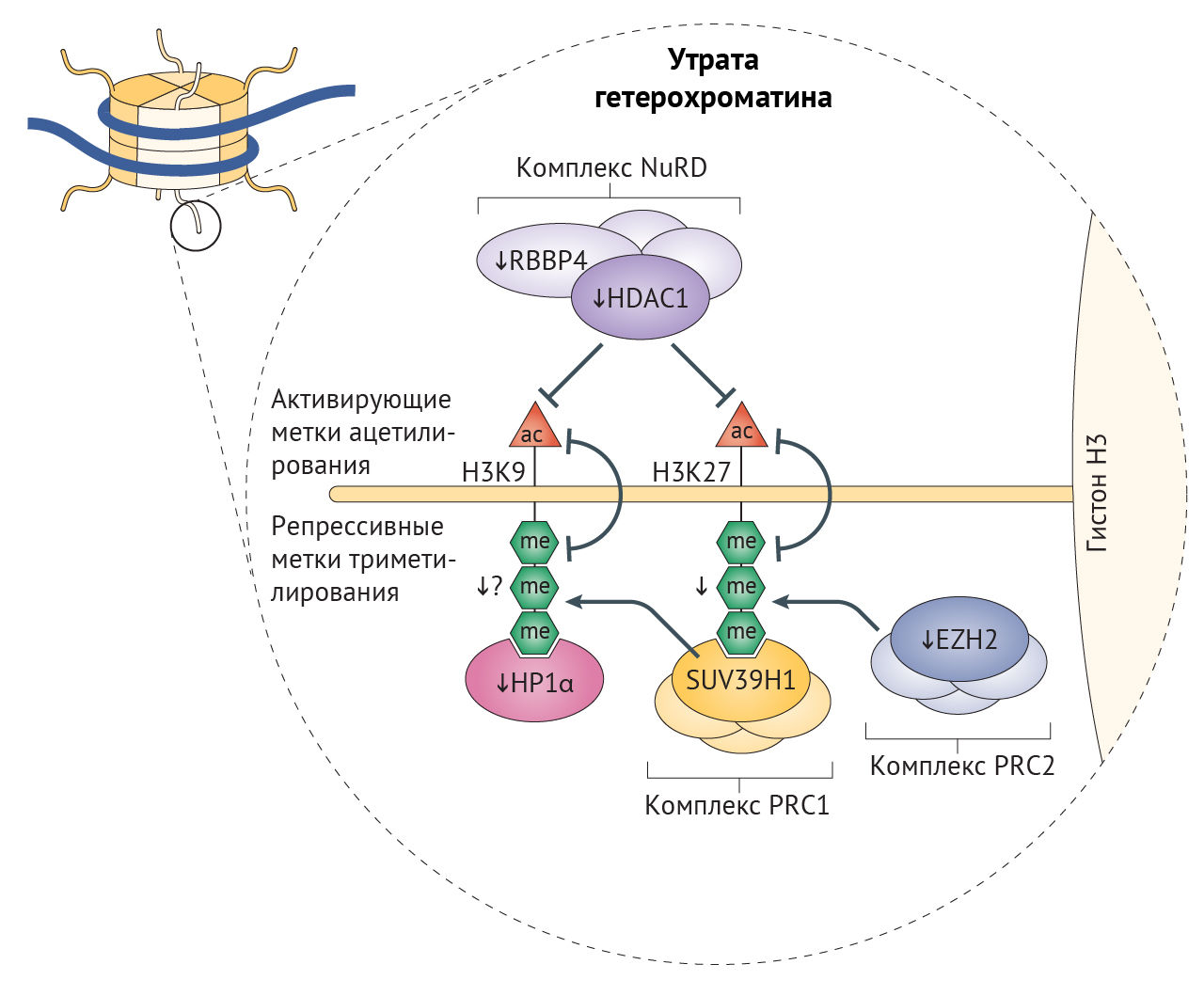
Рисунок 3 | Эпигенетические дефекты, связанные со старением
Общая схема эпигенетических изменений (показано тонкими стрелками) на хвосте гистона Н3, являющихся причиной массового уменьшения доли гетерохроматина при синдроме прогерии Хатчинсона-Гилфорда (СПХГ) и участвующих в старении и развитии болезней старости. Снижение содержания EZH2 приводит к уменьшению триметилирования (показано в виде трех зеленых шестиугольников) гистона Н3 по лизину в 27 положении (H3K27), осуществляемого репрессивным комплексом Polycomb 2 (PRC2) — комплексом белков группы Polycomb (PCG)77, являющегося репрессивной меткой, благодаря которой становится возможным присоединение PRC1 и триметилирование (H3K9me3); к триметилированному гистону может далее присоединиться α-гомолог белка гетерохроматина 1 (HP1α). Это триметилирование снижено при СПХГ [28]. Получены данные, что активность H3K9me3 снижена при СПХГ, но в то же время повышена у мышиных моделей прогерии с нокаутом по Zmpste24 [28, 83]. У больных СПХГ снижена экспрессия различных белков комплекса NuRD (комплекса ремоделирования и деацетилирования нуклеосом) [32], вследствие чего ослабляется деацетилирование гистонов. Предполагается, что разрешающие экспрессию метки ацетилирования по H3K9 и H3K27 (красные треугольники) и запрещающие метки триметилирования по тем же лизинам взаимоисключают друг друга. HDAC1 — гистоновая деацетилаза 1; RBB4 — RB-связывающий белок 4.
Главная функция комплекса PRC2 — стабилизация H3K27me3-меток, с которыми связывается PRC1-комплекс, и индукция транскрипционного сайленсинга в результате стимуляции метилтрансферазы лизина гистонов SUV39H1, обеспечивающей триметилирование лизина H3 в девятом положении (рис. 3) [80]. Нарушения в ядерной ламине влияют на локализацию белков PCG, что приводит к глобальной потере H3K9me3 при СПХГ, а она, вероятно, является причиной дефектов механизмов поддержания длины теломер и аберрантной активации перицентромерных сателлитных повторов, «молчащих» в норме [32,77,81]. Постепенная утрата HP1α, связывающегося с H3K9me3, может также способствовать снижению доли гетерохроматина, что не удается компенсировать с помощью повышения содержания гетерохроматиновой метки H4K20me3 [77]. Постепенная потеря H3K9me3, ведущая к недостатку белка Вернера в человеческих клетках, вероятно, является результатом уменьшения активности SUV39H1 и связывания HP1α [82]. В то же время у мышиной модели прогерия-подобного фенотипа с делецией фермента ZMPSTE24, осуществляющего процессинг преламина А, наблюдалось повышение H3K9me3. У этой модели утрата SUV39H1 приводила к восстановлению целостности генома и увеличивала срок жизни [83]. Снижение метилирования H3K9 также связано с некоторыми AADs, а при ХПБ вызывает изменения экспрессии генов, характерные для сосудистого воспаления [84]. При семейной болезни Альцгеймера из-за накопления мутантного тау-белка снижаются уровни H3K9me2 и HP1α, результатом чего является повышение экспресии нейротоксичного piwi-подобного белка 1 (PIWIL1) [85]. Более того, снижение экспрессии HP1α встречается во многих злокачественных опухолях, коррелируя с неблагоприятностью прогноза [86].
Массовое снижение метилирования гистонов при СПХГ сопровождается гипоацетилированием гистонов H2B и H4, обусловленным, возможно, ослаблением связи гистон-ацетилтрансферазы KAT8 с ядерной ламиной [87]. Примечательно, что как оверрэкспрессия KAT8, так и лечение ингибиторами гистон-деацетилазы (HDAC) продлевают жизнь нокаутированным по Zmpste24 мышам с прогерия-подобным фенотипом [87]. Метка ацетилирования H4K16 (H4K16Ac), связанная с активацией транскрипции и ее репрессией, способствует НСК и гомологичной рекомбинации; а повышение содержания метки продлевает срок жизни клетки [88, 89]. Терапия ингибиторами HDAC способна предотвратить связанное со старением ухудшение когнитивных функций и уменьшить тяжесть ишемического инсульта, болезни Паркинсона и остеопороза у мышиных моделей благодаря изменению экспрессии генов, способствующих и препятствующих болезни [90,91]. HDACs, включая сиртуины, регулируют ацетилирование и негистоновых белков, в число которых входят мастер-регуляторы клеточного роста и метаболизма, на основании чего можно сделать вывод о непосредственной роли эпигенетической регуляции метаболического контроля старения [26].
Метаболический сигналинг критически важен для поддержания клеточного гомеостаза, т. к. благодаря ему распределяется энергия в пользу необходимых защитных механизмов, стоящих на страже целостности генома, эпигенома, протеома и органелл. Его дизрегуляция, запускающая каскады разрушительных событий в клетке, оказывает большое влияние на патогенез AADs.
Обеспечение надлежащего баланса между катаболическими и анаболическими путями необходимо для поддержания адекватного уровня энергии в клетке. Нарушение этого равновесия вызывает сбои в клеточном гомеостазе и ведет к старению клетки и развитию AADs [26]. Нутриент-чувствительные сигнальные пути, включая пути инсулино-подобного фактора роста 1 (ИФР-1) и инсулина (IIS), а также сиртуина 1 (SIRT1) и АМФ-активируемой протеинкиназы (АМФК), регулируют метаболический статус клеток и играют заметную роль в физиологическом старении [26].
Дизрегуляция сигналинга ИФР-1 является причиной старения. Сигнальный путь IIS активируется в условиях избытка нутриентов, к коим относятся инсулин, ИФР-1 и свободные аминокислоты. Это приводит к активации пути mTOR (рис. 4), а различные эффекторы на последующих этапах каскада опосредуют повышение синтеза белка и активируют другие анаболические процессы [92]. Снижение IIS-сигналинга, обусловленное генетическим полиморфизмом или же недостатком калорий, способствует долголетию и здоровью [26]. Тем не менее эти эффекты зависят от возраста организма, длительности и степени репрессии IIS, а также от интенсивности клеточных стрессоров, таких как воспаление. В то время как кратковременная и умеренная репрессия IIS может оказаться полезной, подавляя клеточный рост ради перераспределения энергии в пользу восстановления клеточных повреждений, долговременная репрессия —- губительна и способствует старению [92]. У мышей с прогерия-подобным фенотипом, нокаутированных по Zmpste24, уровень ИФР-1 резко понижен в течение всей жизни, а терапия ИФР-1 увеличивает продолжительность жизни и отсрочивает проявление признаков прогерии [93]. Болезнь Паркинсона характеризуется длительным ингибированием mTOR, способствующим смерти нейронов; активация же mTOR у животных моделей позволяет частично предотвратить этот процесс [94].
Долговременная активация IIS также может оказывать негативный эффект на здоровье клетки (рис. 4). СД2-индуцированная хроническая активация mTOR инактивирует субстрат инсулинового рецептора 1 (IRS1) с помощью петли обратной связи, отключающей инсулин и рецепторы к ИФР-1 от дальнейшего сигналинга и вызывающей тем самым инсулинорезистентность [94]. Этот механизм лежит в основе возникновения связанных с диабетом симптомов у больных с синдромом Вернера, тяжесть которых может быть уменьшена ингибиторами mTOR [20,95]. Нейроны обладают высокой чувствительностью к изменениям в IIS-сигналинге в силу высокого уровня метаболизма в них и зависимости от глюкозы [94]. У пациентов, страдающих от болезни Альцгеймера, симптомы диабета (причиной которых служат разобщение и неэффективность передачи сигнала рецептором ИФР-1, и которые обостряются при ингибировании инсулиновых рецепторов β-амилоидами) предшествуют когнитивным нарушениям за десятилетия. Лечение рапамицином, ингибитором mTOR, отчасти приводит в норму сигнальный путь инсулин-mTOR и уменьшает дефекты в нейронах животных моделей болезни Альцгеймера [94,96].
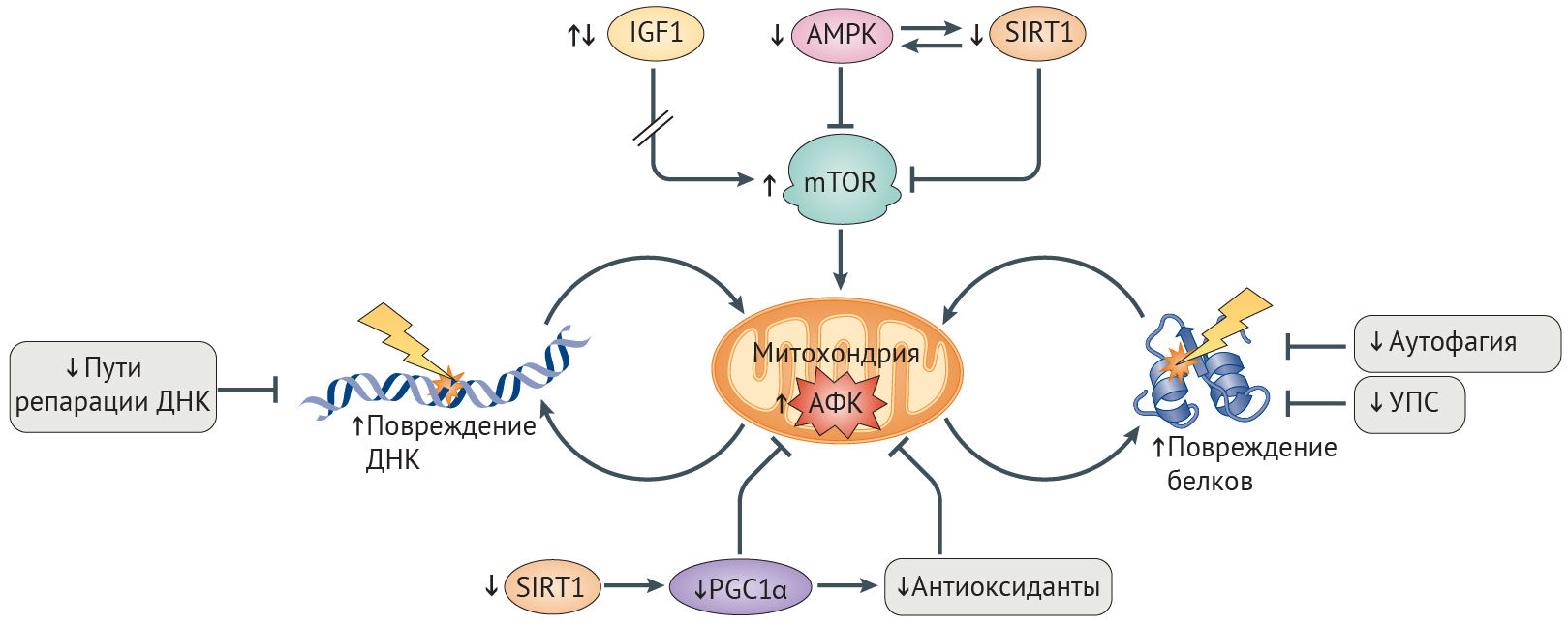
Рисунок 4 | Митохондриальные дефекты старения, вызванные АФК
Дефекты в окислительном фосфорилировании при старении и болезнях, связанных со старением (AADs) обусловлены снижением сигналинга инсулиноподобного фактора роста 1 (ИФР-1), или разобщением путей ИФР-1 и mTOR, следствием чего является постоянная активация пути ИФР-1 (стрелка на рисунке прервана) и повышенная активация mTOR в результате уменьшения активности AMФ-активируемой протенкиназы (АМФК) и сигналинга сиртуина 1 (SIRT1) [26]. Усиление образования активных форм кислорода (АФК) из-за увеличения напряженности работы митохондрий, снижение уровня антиоксидантов и активности PCG1α (коактиватор 1α γ-рецептора, активируемого пролифераторами пероксисом) — всё это приводит к ослаблению биогенеза и обновления митохондрий, к повреждениям ДНК, различных белков и других макромолекул [27]. Неэффективность путей поддержания гомеостаза белков и путей репарации ДНК, включая аутофагию и деградацию поврежденных белков, опосредованную УПС (убиквитин-протеасомной системой), что наблюдается у больных с синдромами преждевременного старения и AADs, еще больше усиливает разрушительный эффект АФК на целостность митохондрий и клеточный гомеостаз [26].
Окислительное фосфорилирование (OXPHOS). Процесс перемещения электронов от доноров электронов к акцепторам, и запасание выделяющейся при этом движении энергии в форме АТФ. У эукариот происходит на внутренней митохондриальной мембране в участках расположения цепи переноса электронов.
Снижение активности сиртуина и АМФК способствует развитию AADs. В условиях стресса и повышенного содержание НАД+ деацетилаза белков SIRT1 активирует метаболические пути, которые повышают уровень энергии в клетке и способствуют ее выживанию [26]. В ходе старения повышенная активность сигналинга повреждений ДНК истощает уровень НАД+, снижая активность SIRT1 (рис. 4) [97]. Важность снижения активности SIRT1 при старении подтверждается наблюдением, что ее восстановление у мышей с прогерия-подобным фенотипом и нокаутом Zmpste24 в результате терапии активатором SIRT1, ресвератролом, приводит к снижению выраженности остеопоротических изменений и увеличению продолжительности жизни [98]. Стимуляция SIRT1 также приводит к облегчению симптоматики болезни Альцгеймера, т. к. при этом ингибируется mTOR, чрезмерно активированный при данной патологии, и индуцируются mTOR-независимые расщепление амилоида и деградация тау-белка (табл. 1) [92].
Повышение активности SIRT1 увеличивает продолжительность здоровой жизни посредством активации АМФК. В ответ на повышение отношения АМФ/АТФ АМФК ингибирует mTOR, стимулирует катаболизм липидов и глюконеогенез и по механизму положительной обратной связи активирует SIRT1 (рис. 4) [99]. Аналогично SIRT1 активность АМФК снижается с возрастом. Соответственно, лечение активатором АМФК, метформином, увеличивает продолжительность жизни C. elegans и мышей, уменьшает выраженность фиброза почек у больных ХБП и оказывает антидиабетический эффект [26, 100]. Метаболические дефекты при СД2 также уменьшаются благодаря усиленной активации SIRT6, возможно путем подавления ИФР-1-сигналинга [92]. У пациентов с СПХГ снижено содержание SIRT6, а оверэкспрессия этого белка несколько уменьшает выраженность старческого фенотипа фибробластов у пациентов с СПХГ и увеличивает срок жизни мышей дикого типа [101,102].
Хотя связь между старением и IIS, АМФК и сиртуин-опосредованным сигналингом очевидна, но то, как именно данные активаторы сигнальных путей влияют на клеточное старение при патологических процессах, известно лишь отчасти. В этом контексте важным фактором, влияющим на клеточное старение, является метаболический контроль целостности митохондрий.
Митохондрии производят АТФ кислород-зависимым методом окислительного фосфорилирования (OXPHOS), путем образования протонного градиента по обе стороны внутренней митохондриальной мембраны (с помощью белковых комплексов цепи переноса электронов), далее используемого АТФ-синтазой. В дополнение к этому, митохондрии обеспечивают регуляцию глюконеогенеза, окисления жирных кислот, уровня внутриклеткочного кальция и апоптоза [103].
Снижение активности OXPHOS при СПХГ, физиологическом старении и AADs. В ходе старения целостность митохондрий нарушается, что проявляется снижением трансмембранного потенциала, повышением образования АФК в процессе синтеза АТФ, дисрегуляцией гомеостаза кальция, индукцией апоптоза и повышением числа мутаций в митохондриальной ДНК (мтДНК, кодирующей белки-участники OXPHOS) [103]. Связь между целостностью митохондрий и старением продемонстрирована с помощью нокина мтДНК-полимеразы POLGα у мышей, которая в результате проявляла сниженную способность к коррекции ошибок репликации. У этих мышей эффективность OXPHOS была уменьшена вследствие накопления мутаций в мтДНК, что являлось причиной ускоренного старения [104]. При СПХГ наблюдается аномальное набухание и фрагментация митохондрий [105], а активность OXPHOS начинает снижаться с раннего возраста [106]. У мышей с нуль-мутацией Wrn митохондрии увеличены, а количество мутаций мтДНК и образование АФК в процессе генерации АТФ повышены [107].
Система OXPHOS является основным поставщиком АФК [108]. Длительное повышение уровня АФК ведет к повреждению клеточных макромолекулярных структур, включая белки комплексов OXPHOS и мтДНК, создавая порочный круг, в котором АФК нарушают целостность митохондрий, что приводит к дальнейшему нарастанию оксидативного стресса (рис. 4) [109]. При болезни Альцгеймера повышенные уровни АФК предшествуют и способствуют образованию β-амилоидных и тау-содержащих бляшек и нейрофибриллярных клубков [103], которые все больше ухудшают работу OXPHOS-комплексов [110]. Подобная самоподдерживающаяся петля наблюдается и при семейной болезни Паркинсона, при которой патогенные белки усиливают образование АФК, снижают эффективность OXPHOS и в конечном счете запускают гибель нейронов [103]. Генетическое выключение TFAM, фактора выживания мтДНК, схожим образом приводит к болезни Паркинсона у мышей [103]. При атеросклерозе постоянное повышение АФК способствует образованию бляшек посредством ингибирования OXPHOS и окисления липопротеинов низкой плотности, которые оказывают свое повреждающее действие, привлекая моноциты [103]. Установлено, что митохондриальная дисфункция индуцирует воспалительные реакции при ХОБЛ [111]. Нарушение целостности митохондрий, вызванное АФК, далее способствует кальцификации сосудов, что наблюдается у лиц с СПХГ, атеросклерозом, остеопорозом, ХБП, СД2 и болезнью Альцгеймера [112,113].
Снижение эффективности антиоксидантной системы и митохондриального биогенеза при СПХГ и AADs. Клетки могут бороться с порочным взаимодействием между АФК и митохондриями путем активации антиоксидантной системы, снижающей уровни АФК (рис. 4). Ядерный фактор эритроид-2-связанного фактора (NRF2) — ключевой регулятор транскрипции антиоксидантов, включая тиоредоксин-зависимую пероксидредуктазу (PRDX3), которая нейтрализует большую часть образующейся в митохондрии перекиси водорода [114,115]. Активность NRF2 снижается с возрастом [116]. Прогерин же связывает NRF2 и тем самым ингибирует его транскрипционную активность, что приводит к хроническому оксидативному стрессу и старению клетки при СПХГ [27]. Дефекты антиоксидантных защитных механизмов лежат в основе болезней Альцгеймера и Паркинсона, что, вероятно, обусловлено либо нарушениями активации NRF2, либо же непосредственным ингибированием антиоксидантов патологическими белками — к примеру, ингибирование PRDX3 мутантным белком LRRK2, приводящим к болезни Паркинсона [108,115,117]. Восстановление митохондриальной целостности посредством оверэкспрессии каталазы — митохондрия-адрессованного антиоксиданта — увеличивает продолжительность жизни мышам и оказывает протективное действие в отношение нейродегенерации, болезней сердца, онкологических заболеваний и инсулинорезистентности [103,118].
Усиленное обновление митохондрий и замещение поврежденных митохондрий интактными также защищает от оксидативного повреждения. Коактиватор-1α γ-рецептора, активируемого пролифераторами пероксисом (PGC1α) способствует митохондриальному биогенезу, а параллельно этому активирует NRF2-опосредованный антиоксидантный ответ (рис. 4) [103]. Активация PGC1α уменьшает тяжесть митохондриальной дисфункции и других дефектов старения в фибробластах пациентов с СПХГ [105], несмотря на то, что, судя по всему, положительные эффекты активации PGС1α in vivo являются тканеспецифичными и зависят от длительности и степени активации [37]. Сниженная активность PGС1α наблюдается при болезнях Альцгеймера и Паркинсона и сопричастна развитию связанной с ХБП мышечной дистрофии [110,119]. Наблюдаемое при СПХГ, старении и AADs снижение активности PGC1α вероятнее всего и объясняется недостаточной активацией SIRT1 и АМФК (вышестоящих регуляторов PGС1α) [26].
Таким образом, эти наблюдения показывают, что метаболический сигналинг связывает между собой митохондриальную целостность и образование АФК. Баланс в этой триаде оказывает большое влияние на протеолитическую систему аутофагии, участвующую в деградации дефектных митохондрий, обороте аминокислот, требуемых для регулируемого mTOR синтеза белков, и элиминации поврежденных АФК макромолекул в целях поддержания гомеостаза клетки.
Под протеостазом понимается поддержание функционального протеома путем сбалансированной регуляции синтеза белка, восстановления его структуры и протеолиза. Целостность протеома зависит от беспрерывного обновления белков, заключающегося в деградации поврежденных и неверно уложенных пептидов посредством аутофагии и убиквитин-протеосомной системы (УПС), а при прогероидных синдромах человека, физиологическом старении и AADs нарушается и то, и другое.
Убиквитин-протеасомная система (УПС). Комплекс, обеспечивающий АТФ-зависимую деградацией белков, помеченных специальной меткой — убиквитином.
Метаболически обусловленное накопление поврежденных митохондрий и агрегатов окисленных белков при старении и при патологиях говорит о дисбалансе между действием протеотоксических факторов и протеолитических путей, уничтожающими поврежденные органеллы и белковые агрегаты. Аутофагия является главным механизмом деградации, при котором аутофагосомы поглощают поврежденные компоненты клетки и способствуют их деградации посредством слияния с лизосомами, содержащими гидралазу [120].
Любопытно, что каскад IIS ингибирует аутофагию путем активации mTOR (рис. 4) [26]. Тогда как временная приостановка аутофагии, вызванная mTOR, может быть полезна для здоровья клетки, способствуя анаболизму, длительное ингибирование аутофагии приводит к накоплению токсичных белковых агрегатов в тельцах включения, т. н. агресомах, устойчивых к деградации. Агресомы содержат адапторный белок аутофагии p62 (известный также как SQSTM1) и убиквитин, которые в норме притягивают белковые агрегаты и способствуют их протеолизу [120]. При СПХГ были обнаружены убиквитин- и p62-положительные агрегаты прогерина [121]. Считается, что прогериновые агрегаты вредны для клетки из-за способности включать в свой состав нормальные клеточные белки, в том числе и NRF2 [27]. Активация аутофагии путем рапамицин-опосредованного ингибирования mTOR способствует повышению растворимости и деградации прогерина, тем самым уменьшая тяжесть дефектов при клеточном старении [121]. Терапия рапамицином увеличивает продолжительность жизни как у мышей, так и у C. elegans; причем этот эффект, вызванный ингибированием IIS, зависим от аутофагии [26,122].
Существует доказательства положительного влияния стимуляции аутофагии при AADs, и эти эффекты большей частью можно свести к повышению утилизации патогенных токсических агрегатов, образующихся под действием хронического оксидативного стресса [120]. При болезни Альцгеймера уровень лизосомальной протеазы, катепсина D, снижен, из-за чего нарушается деградация β-амилоида и агрегатов фосфорилированного тау-белка. Ослабление гиперактивированного сигналинга IIS-mTOR повышает интенсивность аутофагии этих агрегатов и уменьшает выраженность нарушений памяти у мышиных моделей болезни Альцгеймера [123,124]. У лиц с болезнью Паркинсона снижены уровни лизосом-ассоциированного мембранного гликопротеина 2 (LAMP2), который, предположительно, препятствует протеолизу α-синуклеиновых агрегатов и способствует смерти нейронов [125]. Вдобавок к этому, генетические дефекты в белках parkin и PTEN-индуцированной киназы 1 (PINK1) (см. Вставка 1) снижают активность аутофагии поврежденных митохондрий при семейной болезни Паркинсона [120].
Дефицит LAMP2A и катепсина D является также известной причиной кардиомиопатий, а ослабление аутофагии может способствовать атеросклерозу вследствие активации клеточной смерти и старения ГМС [126,127]. Агрегация белков также участвует и в патогенезе СД2: было обнаружено, что в поджелудочной железе у мышиных моделей СД2 образуются агрегаты амилина; более того, агрегация усиливается при генетической абляции белка аутофагии ATG7 [128]. Кроме этого мутация, повышающая способность амилина образовывать агрегаты, также ассоциирована с СД2. Агрегация мутантного амилина предшествует развитию дисфункции панкреатических β-клеток и этого достаточно, чтобы индуцировать диабетический фенотип у крыс дикого типа [128].
УПС обеспечивает деградацию поврежденных белков путем АТФ-зависимого протеолиза [126]. У пациентов с СПХГ ее активность снижена (рис. 4) [129], и она так же снижается в ходе старения. С этим согласуются полученные данные о том, что повышенная активность УПС связана с увеличением продолжительности жизни Drosophila melanogaster и C. elegans [129].
Дофаминэргическая абляция субъединицы P26S4 УПС у мышей приводит к усилению агрегации α-синуклеина и к гибели нейронов, подобно тому, как это происходит при болезни Паркинсона [120]. Вдобавок к этому, пониженные уровни PA700 и PA28, активаторов УПС, в черном веществе коррелируют со степенью агрегации α-синуклеина у пациентов с болезнью Паркинсона [130]. Умеренное снижение активности УПС путем оверэкспрессии мутантной доминантно-негативной каталитической субъединицы протеасом PSMB5 усугубляет поражения сердца, вызванные ишемией-реперфузией, а повышение уровня пре-амилоидных олигомеров сопровождает дилатационную и гипертрофическую кардиомиопатии [126]. Соответственно, стимуляция УПС — перспективная терапевтическая стратегия для лечения кардиомиопатий [126]. Таким образом, данные наблюдения говорят нам о том, что аутофагия и УПС играют ключевую роль в противодействии связанному со старением метаболически индуцированному стрессу.
Аккумуляция клеточных повреждений при старении приводит к запуску сигнальных путей, контролирующих пролиферацию и дифференцировку клеток, равно как и опосредованную стволовыми клетками регенерацию, в целях предупредить постоянный коллапс гомеостаза на клеточном и тканевом уровнях. Особенно важное место в старении и развитии AADs занимает клеточное старение и пути регенерации.
Потеря геномной и протеомной целостности может запускать клеточное старение, являющееся, по сути, необратимым арестом пролиферации, главным образом зависимым от путей супрессии опухоли RB-p16 и p53-p21 (рис. 5) [131,132]. Временная активация этих путей может иметь позитивное значение, поскольку она позволяет восстановить клеточные повреждения прежде, чем продолжится пролиферация. Тем не менее, длительная активация RB-p16 и p53-p21 приводит к негативным последствиям, потому что она запускает самоподдерживающийся воспалительный сигналинг, обусловленный трансформирующим фактором роста-β (ТФР-β) и ядерным фактором каппа-B (NF-κB). Этот сигналинг характеризуется экспрессией интерлейкина-1 (ИЛ-1) и ИЛ-6, фактора некроза опухоли-α (ФНО-α), циклооксигеназы 2 (ЦОГ-2) и хемокином с мотивом C-X-C 1 (CXCL1) и CXCL2 (рис. 5) [131]. Это состояние получило название связанный со старением секреторный фенотип (SASP), и долговременная активация данного сигналинга может нарушить коммуникацию между стареющими и иммунными клетками, тем самым предотвращая опосредованную иммунной системой элиминацию стареющих клеток, которая в норме и запускается SASP [131]. Невозможность уничтожения стареющих клеток может еще больше усугубить возрастное нарушение физиологических функций, индуцируя клеточное старение в соседних клетках посредством щелевых контактов [26]. Не противоречит этому наблюдение, что искусственно вызванная элиминация стареющих клеток увеличивает продолжительность и качество жизни прогероидных мышей с отсутствием белка контрольной точки митоза BubR1 (табл. 1), препятствует атерогенезу у мышей с дефектом рецептора к липопротеинам низкой плотности и оказывает протективное действие на почки и сердце у мышей дикого типа [6,7,133].
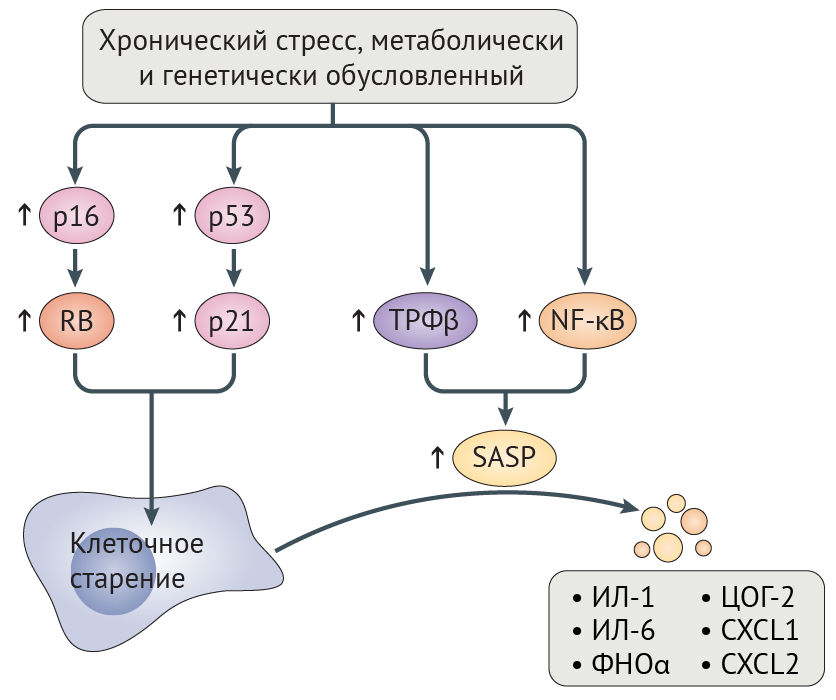
Рисунок 5 | Пути клеточного старения
Хронически повышенный уровень повреждения ДНК и метаболического стресса при синдроме прогерии Хатчинсона-Гилфорда (СПХГ), физиологическом старении и болезнях старости (показано стрелками) запускает активацию р16 и р53. Последние активируют RB и p21, что приводит к перманентной остановке роста клетки [26]. Повышенная активность трансформирующего фактора роста-β (ТФР-β) и ядерного фактора каппа B (NF-κB) ведет к формированию старческого секреторного фенотипа (SASP), который характеризуется наличием показанных на рисунке эффекторов воспаления.
ЦОГ-2 — циклооксигеназа 2; CXCL1 — хемокин с мотивом C-X-C 1; ФНО-α — фактор некроза опухоли-α.
При СПХГ увеличенное количество клеточных повреждений приводит к гиперактивации RB-p16, p53-p21 и NF-κB, своим следствием имея клеточное старение и SASP, что сопровождается повышением уровней ИЛ-6, ФНО-α, CXCL1 и CXCL2 у мышиных моделей прогерии [34,134,135]. Гиперактивация NF-κB отчасти вредна из-за эпигенетических эффектов, поскольку это активирует гистон-метилтрансферазу DOT1L [136]. И наоборот, инактивация NF-κB или p53 на генетическом уровне увеличивает продолжительность и качество жизни у мышиных моделей СПХГ [34,134].
При болезни Альцгеймера поддерживающие нейроны астроциты, которые обладают естественной высокой базальной активностью NF-κB и вовлечены в патогенез данного заболевания, демонстрируют повышенную экспрессию p16 и ИЛ-6 [137]. Гиперактивация NF-κB по каноническому пути пагубна для астроцитов и, судя по всему, способствует агрегации β-амилоида, в свою очередь обратимо запускающего опосредованные ФНО-α, ИЛ-1β и ЦОГ-2 воспалительные сигнальные каскады [138]. Болезнь Паркинсона, аналогично болезни Альцгеймера, ведет к повышению активности NF-κB и уровней ЦОГ-2 [139]. Терапия мышей дикого типа ингибиторами ЦОГ-2 нивелирует нейротоксический эффект МФТП (1-метил-4-фенил-1,2,3,6-тетрагидропиридина), инициирующего болезнь Паркинсона посредством ингибирования OXPHOS [139]. Повышенные уровни ИЛ-1β далее усугубляют прогрессирование болезни Паркинсона, индуцируя экспрессию α‑синуклеина [139].
При старческом идиопатическом фиброзе легких повышение содержания ИЛ-1β может активировать дифференцировку фибробластов легких в миофибробласты, тем самым поддерживая фиброз [140]. SASP-регулируемые изменения клеточных характеристик играют роль и в злокачественных процессах, в силу того, что ИЛ-6 и ИЛ-8 вносят вклад в развитие опухолей, содействуя эпителиально-мезенхимальному переходу (ЭМП) [131]. Ингибиторы ТФР-β, являющиеся супрессорами SASP, продемонстрировали способность подавлять прогрессию некоторых опухолей и в настоящее время проходят клинические испытания [141]. Наконец, ТФР-β- и NF-κB-обусловленные воспалительные ответы, ведущие к SASP, участвуют в патогенезе ХБП, и репрессия обоих путей ингибиторами ангиотензин-превращающего фермента замедляет развитие ХБП [141].
Эпителиально-мезенхимальный переход (ЭМП). Процесс, в ходе которого эпителиоциты претерпевают различные изменения на молекулярном уровне: изменяется характер межклеточных контактов, полярности клетки и способность к инвазии, с целью превращения в мезенхимальные клетки. ЭМП играет положительную роль при заживлении ран, но в то же время и негативную — при фиброзе внутренних органов и инициации метастазирования опухолей.
В целом, необратимая остановка роста и устойчивая гиперактивация воспалительных секреторных сигнальных путей ведут к перманентному состоянию клеточного старения, лежащего в основе старения и AADs. Накоплению стареющих клеток может препятствовать регенерация посредством стволовых клеток.
Стволовые клетки, которые способны к самообновлению и мультилинейной дифференцировке, располагаются в специфических нишах по всему человеческому телу. Человеческие мезенхимальные стволовые клетки (МСК), присутствующие фактически во всех тканях, дифференцируются в остеоциты, хондроциты и адипоциты, в то время как ГМС, не окончательно дифференцированные клетки сосудистой стенки, обладают ограниченным количеством путей дифференцировки [142,143]. Связанные со старением дефекты фибробластов, взятых от пациентов с СПХГ, могут быть обращены вспять путем преобразования фибробластов в индуцированные плюрипотентные стволовые клетки (иПСК), не экспрессирующие прогерин из-за естественной репрессии транскрипции гена LMNA [30, 31]. После повторной дифференцировки СПХГ-иПСК в МСК и ГМС реактивация транскрипции гена LMNA и, соответственно, индукция экспрессии прогерина ведет к появлению типичных геномных, протеомных и клеточных дефектов старения, характерных для СПХГ (рис. 1–4) [30,31], и снижает выживаемость при таких стрессовых состояниях как гипоксия. Эта зависимость может объяснить потерю ГМС у пациентов с СПХГ при гемодинамическом стрессе, равно как и сниженную способность СПХГ-МСК предотвращать повреждения в ходе ишемии-реперфузии на мышечной модели задней конечности [31]. Выживаемость и способность СПХГ-МСК противостоять хроническому оксидативному стрессу может быть восполнена посредством повторной активации NRF2, ингибируемого прогерином [27].
В дополнение к истощению пула стволовых клеток, которое также может быть характерно для стволовых клеток дермы у мышиной модели СПХГ, накопление дефектов старения может нарушать регенеративную способность стволовых клеток путем изменения их потенциала дифференцировки [144]. Так прогерин задерживает дифференцировку ГМС и способствует остеогенной дифференцировке МСК за счет усиленного образования адипоцитов, что может способствовать кальцификации тканей (таких как кожа и стенки сосудов), наблюдаемой у пациентов с СПХГ [48,145].
Наблюдаемое у стареющих пациентов накопление небольших количеств прогерина и малое содержание белка Вернера в ГМС и МСК дикого типа, соответственно, также может объяснить снижение регенеративной способности стволовых клеток в ходе физиологического старения [82,146]. Действительно, сообщается, что в ходе старения в МСК, ГМС, гемопоэтических стволовых клетках (ГСК), клетках скелетных мышц и нервных стволовых клетках наблюдаются один или более типичных признаков клеточного старения, таких как оксидативный стресс или активация сигнальных путей повреждения ДНК, старения и SASP [143,147]. Негативные эффекты этих признаков на стволовые клетки наглядно проявляются ухудшением восстановления тканей легких и поджелудочной железы, в норме обладающих высоким регенеративным потенциалов, у пожилых людей после частичной резекции — по сравнению с молодыми пациентами [148, 149]. Таким образом, сайленсинг p16, стимуляция SIRT1, блокирование эффектов SASP путем оверэкспрессии антагониста рецептора ИЛ-1 или же уменьшение образование АФК путем оверэкспрессии антиоксиданта, супероксиддисмутазы 2, повышают регенеративную способность ГМС и ГСК, равно как и нервных, кишечных и мышечных стволовых клеток [143,147].
Взрослые стволовые клетки могут быть более подвержены возрастным дефектам, т.к. они в основном находятся в состоянии покоя (т. е. в состоянии временного ареста пролиферации) чему, по некоторым данным, сопутствуют снижение уровня белков репарации ДНК и отсрочка репарации ДНК до момента вступления в новый клеточный цикл [147]. По всей видимости, накопление повреждений ДНК во время этого периода покоя может пересечь ту пороговую черту, после которой начинается снижение функции стволовой клетки.
Снижение регенеративной способности стволовых клеток играет важную роль при некоторых AADs. К примеру, старение ГМС ведет к образованию атеросклеротических бляшек [150]. При СД2 МСК могут обладать сниженной способностью к пролиферации и дифференцировке, а активность МСК и панкреатоспецифических стволовых клеток играет важную роль в поддержании количества и функции β-клеток поджелудочной железы [148]. Более того, потенциал дифференцировки стволовых клеток легких нарушается при старческом идиопатическом легочном фиброзе, а функциональные МСК способствуют заживлению остеопоротических переломов [143,149]. Хотя и неизвестно, участвуют ли сердечные и нервные стволовые клетки в обновлении окончательно дифференцированных миоцитов и нейронов [147,151], их позитивные секреторные иммунорегуляторные эффекты установлены, а на мышиных моделях болезни Альцгеймера было продемонстрировано, что после инъекции МСК смягчаются когнитивные нарушения [147,151].
Таким образом, на основе этих данных о болезнях преждевременного старения, физиологическом старении и AADs можно предположить, что преждевременное старение и AADs обусловлены генетическими, метаболическими дефектами и дефектами клеточного старения, которые вызывают сдвиги в судьбе стволовых клеток.
В данной работе авторы проанализировали связь между преждевременным старением, нормальным старением и AADs. Обсудили, как каждый конкретный дефект в регуляции геномной, теломерной, эпигенетической, метаболической, митохондриальной и протеолитической целостности, а также целостности клеточного цикла, влияет на процесс развития клеточного старения и AADs (рис. 1). Представленный рисунок отображает одну из общих схем связи между этими ключевыми признаками старения; как можно видеть, нарушение одного из этих процессов вызывает появление дефектов в других, что в конечном счете ведет к коллапсу клеточного гомеостаза, обуславливающему болезнь. Хотя этот каскад уменьшает продолжительность здоровой жизни и запускает старение, тесное переплетение этих событий может предоставить возможностью лечения: терапии, направленной на один (или более) из этих признаков, может оказаться достаточно для облегчения процесса в целом.
Наблюдения, полученные в исследованиях систем преждевременного старения, нормального старения и AADs дополняют друг друга. Хотя прогероидные синдромы отражают только некоторые аспекты процесса старения, тем не менее, они представляют немалый интерес по причине четко детерминированных генетических признаков и возможности проведения контролируемых экспериментов.Физиологическое старение и AADs отражают действительный процесс старения, но их изучение сопряжено с трудностями интерпретации в контексте физиологии и среды. Чтобы дальше распутать эту, богатую тонкостями, сеть клеточного старения, потребуется разработка новейших экспериментальных модельных систем, которым не будет помехой традиционно медленное стохастическое накопление и низкая пенетрантность дефектов старения. Прогероидные синдромы у человека предоставляют такую возможность в силу того, что связанные дефекты клеточного старения во многом повторяют те, что наблюдаются при физиологическом старении и AADs. Более того, ускоренная природа патогенеза болезней преждевременного старения, их четко определяемая генетическая причина и их мощнейшая фенотипическая пенетрантность позволяют разработать надежные методы изучения клеточного старения.
Создание иПСК, полученных от пациентов с этими и другими болезнями преждевременного старения — новая многообещающая стратегия, дающая возможность в реальном времени наблюдать формирование уже известных возрастных дефектов [152]. В качестве альтернативы, индукция экспрессии мутантных белков, связанных с прогероидными синдромами в клетках дикого типа, может тоже ответить на эти вопросы, а обе стратегии хороши еще и тем, что их легко можно комбинировать с доступными высокопроизводительными скрининговыми методами РНК-интерференции (RNAi) и CRISPR. Использование системы редактирования генома CRISPR-Cas9 может ускорить описание недавно идентифицированных механизмов старения, способствуя созданию in vivo новых моделей старения и моделей AADs.
Авторы ожидают, что разработка новых экспериментальных методик для изучения болезней преждевременного старения человека начнет отвечать на ключевые вопросы о том, каким образом дефекты клеточного старения связаны между собой и как они взаимодействуют с путями, противодействующими старению. Подобные исследования улучшат понимание процесса старения и облегчат диагностику и лечение ведущих хронических заболеваний, связанных со старением, таких как болезни Альцгеймера и Паркинсона, атеросклероз, диабет и рак.
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Статья на конкурс «био/мол/текст»: Эпигенетика — относительно недавно возникшая область науки. Предметом ее изучения являются устойчиво сохраняющиеся в ряду клеточных делений изменения активности генов, не связанные с изменением самой ДНК. Иными словами, за счет эпигенетических модификаций одни гены работают, а другие — молчат. Еще тридцать лет назад в научном сообществе многие из-за укоренившихся догм не хотели признавать важность эпигенетических процессов в биологическом мире. Сейчас к этой области знания приковано внимание множества лабораторий и институтов по всему миру, занимающихся в том числе и изучением природы старения. В геронтологии получило мощное развитие новое направление, связанное с описанием эпигенетических механизмов возрастных изменений.
Эпигенетика начинает поиск
Эпигенетика — это научное направление, изучающее все факторы, которые влияют на активность генома, но не связаны с мутациями ДНК. Так как все наши клетки содержат одинаковую ДНК, эпигенетическое управление активностью генов имеет первостепенное значение при установлении специализации клеток во время развития организма и в последующем функционировании генома. Эпигенетика посредством своих механизмов — например, метилирования цитозиновых оснований ДНК, модификации гистонов и РНК-интерференции (рис. 1) — управляет работой генома, изменяя структуру хроматина и «включая» или «выключая» наши гены .
Подробнее о процессах эпигенетической модификации генома — в статьях: «Развитие и эпигенетика, или история о минотавре» [1], «Эпигенетические часы: сколько лет вашему метилому?» [2], «Эпигенетика поведения: как бабушкин опыт отражается на ваших генах» [3].
Метилирование представляет собой способ регулирования активности генов путем присоединения к цитозиновым основаниям ДНК метильной группы (-СН3). Метилирование подавляет активность гена: синтез РНК (соответственно, и белка) по такой матрице становится невозможным. Это своего рода «заглушка», которую организм использует, инактивируя те или иные гены, работа которых в данный момент ему не нужна или может представлять опасность.
Еще одним важным механизмом эпигенетической регуляции является модификация гистонов, входящих в состав хроматина. Хроматин — это комплекс белков и нуклеотидов, обеспечивающий надежное хранение и нормальную работу ДНК. В наших клетках упаковка ДНК похожа на склад бижутерии. Иначе никак невозможно уложить спираль длиной два метра в одно маленькое клеточное ядро. Нить ДНК наматывается в полтора оборота на многочисленные «бусинки» из нескольких специальных белков, гистонов. Так формируются повторяющиеся структурные единицы эукариотического хроматина, называемые нуклеосомами. В каждой нуклеосоме содержится восемь молекул гистонов — по две молекулы каждого из четырех видов белков (H2A, H2B, H3, H4). Гистоны имеют «хвостики» — белковые «наросты», которые могут удлиняться или укорачиваться специальными ферментами. Длина такого «хвоста» напрямую влияет на активность генов, находящихся вблизи него.
Гистоновый «хвост» может подвергнуться процессу ацетилирования, то есть замене в нём атомов водорода на остаток уксусной кислоты. В результате этого связь гистонов с ДНК, основанная на притяжении разнозаряженных частиц, ослабится, гистон диссоциирует (отдалится от ДНК), и упаковка ДНК станет более «рыхлой» (менее плотной). Тогда белки-регуляторы смогут легче присоединяться к ДНК, и активность гена повысится. Эпигенетические процессы взаимосвязаны друг с другом: так, увеличение уровня метилирования ДНК обычно сопряжено со снижением уровня ацетилирования гистонов, и наоборот. Помимо ацетилирования существует множество других способов модификаций гистонов: метилирование, фосфорилирование, сумоилирование и так далее. Эти модификации могут как разрыхлять, так и «заматывать крепче» упаковку ДНК, что приводит к увеличению или уменьшению экспрессии генов соответственно.
Не так давно был обнаружен еще один уровень эпигенетической регуляции, заключающийся в воздействии на экспрессию генов посредством некодирующих РНК (нкРНК). Эти РНК могут регулировать активность гена как на транскрипционном, так и на посттранскрипционном уровнях — то есть могут как «заглушать» транскрипцию, предотвращая синтез мРНК и препятствуя тем самым считыванию информации с гена, так и приводить к разрушению уже синтезированной мРНК. Этот механизм назвали РНК-интерференцией. В 2006 году за его открытие американские исследователи Эндрю Файер и Крейг Мелло были удостоены Нобелевской премии в области физиологии и медицины .
Подробнее о РНК и связанных с ней механизмах воздействия на геном рассказано в статьях: «Обо всех РНК на свете, больших и малых» [4], «Как избавиться от РНК за несколько минут» [5], «микроРНК — чем дальше в лес, тем больше дров» [6].
Важные научные открытия, связанные с эпигенетикой, появляются практически ежегодно. И хотя ученые находятся в самом начале пути, полученные данные показывают исключительную ценность этого научного направления. Получив мировое признание, эпигенетика вскоре перешла из чисто научной области исследований в очень прикладную, когда выявилась ее тесная связь с развитием и старением. Не случайно одна из недавних статей в Nature вышла под названием «Эпигеномика начинает поиск» [7]. И этот поиск уже дает свои результаты.

Рисунок 1. Структура хроматина и эпигенетические механизмы воздействия на геном. Модификации гистонов: Ас, ацетилирование; Me, метилирование; Ub, убиквитинирование; P, фосфорилирование. Метилирование цитозина ДНК — Me ~ C. РНК-интерференция: miРНК, малые некодирующие РНК.
Метилирование и его связь со старением
В научных трудах по геронтологии часто встречается описание онтогенеза (индивидуального развития организма) лососевых рыб. И, как уже известно, молниеносно развивающееся сразу после нереста старение рыб этого вида сопровождается массивным деметилированием ДНК [8]. Установлено, что с возрастом происходит общее снижение уровня метилирования ДНК. В ДНК, которую брали у эмбрионов и новорождённых, присутствует наибольшее количество метилированных цитозиновых оснований. Получается, что некоторые гены, которые были «заглушены» и молчали в детском и молодом возрасте, к старости начинают проявлять активность. Другая, меньшая часть генов, напротив, с возрастом «замолкает», подвергшись метилированию. Последствия таких изменений генной активности сегодня еще не до конца изучены, но некоторые аспекты уже известны.
Так, довольно большая часть метилированного генома человека (по некоторым данным, до 90%) приходится на подвижные элементы ДНК — ретротранспозоны . Некоторые вирусные агенты, такие как аденовирус или вирус гепатита В, попадая в наш организм, также могут блокироваться посредством метилирования. Характерный для человека ретротранспозон Alu из-за ослабления его метилирования в старости может начать перемещаться — создавать свои копии и вставлять их в разные точки генома, нарушая этим нормальную работу генов. Подобные неконтролируемые перемещения ретротранспозонов несут в себе немалую опасность и могут быть причиной серьезных патологий: сегодня с активностью подвижных элементов ДНК связывают около ста заболеваний [9].
О мобильных генетических элементах эукариот можно прочитать в статьях: «Тайны „молекулярных паразитов“, или Как путешествовать по геному» [10], «Разнообразия много не бывает: чем занимаются мобильные элементы генома в мозге» [11], «Геном человека: полезная книга, или глянцевый журнал?» [12].
Насколько большим может быть влияние эпигенетики на продолжительность жизни, показали в своих работах ученые из Австралийского национального университета, Роберт Кухарски и его коллеги. В 2008 году Science опубликовал результаты их исследований о влиянии фермента ДНК-метилтрансферазы-3 (DNMT-3) на продолжительность пчелиной жизни [13]. Долгое время оставалось загадкой, каким образом из генетически совершенно одинаковых личинок появляются две разные касты пчел — рабочие и королевы (или матки).

Рисунок 2. Комплекс ДНК-метилтрансферазы с ДНК.
Если рабочие пчелы живут всего несколько недель, то матки — несколько лет. Такая огромная разница в длине жизненного пути генетически одинаковых организмов является следствием особого питания: тех личинок, которым суждено стать королевами, дольше кормят маточным молочком. Молекулярные механизмы этого явления стали понятны, когда Р. Кухарски и его команда искусственно уменьшили количество фермента DNMT-3 у личинок пчел. Этот фермент прикрепляет метильные группы к ДНК (рис. 2), подавляя экспрессию генов. Без DNMT-3 активность некоторых генов у личинок оказалась повышенной, и в итоге большинство из них превратилось в королев даже без кормления маточным молочком. Расшифровка пчелиного эпигенома подтвердила это предположение: в ДНК пчеломаток было найдено значительно меньше метильных групп, чем у рабочих пчел. Несколькими годами позже группа Р. Кухарски, проводя исследование уровня метилирования цитозина в нервной системе рабочих пчел и пчеломаток, нашла 561 ген со значительными различиями в метилировании между двумя пчелиными группами [14].
Конечно, не только особенности эпигенома влияют на срок жизни пчеломатки. Как сегодня известно, долгожительницу из королевы делает совокупность факторов: это и нужное соотношение полиненасыщенных и насыщенных жирных кислот, и уменьшенное содержание цитохрома с (который способен запускать процесс клеточной гибели, апоптоз), и повышенная антиоксидантная активность [15].
Как выяснилось, влияние эпигенома на продолжительность жизни чрезвычайно велико и у людей. Так, в 2013 году большая группа итальянских генетиков, возглавляемая Джованни Витале, опубликовала результаты работы, в ходе которой изучались возрастные изменения метилирования ДНК [16]. Объектами исследования стали две группы женщин-ровесниц, жительниц Северной Италии. В одной группе были собраны пожилые итальянки, имевшие матерей-долгожительниц и отцов, проживших не менее 77 лет. К другой группе отнесли итальянок, родители которых умерли, прожив около 70 лет (отцы — 67 лет, матери — 72 года). Распределив таким образом исследуемых, ученые поставили себе задачу сравнить, какие изменения на генном уровне могут лежать в основе долголетия. А также выяснить, существует ли явная преемственность в этом вопросе — передаются ли факторы долгожительства по наследству?
Результаты их работы превзошли все ожидания и показали следующее.
Снижение метилирования ДНК (гипометилирование), характерное для пожилого возраста, происходило гораздо быстрее у итальянок, чьи родители не дожили до 70 лет, чем у их сверстниц, имевших родителей-долгожителей. Исследователи обнаружили, что метилирование (а значит, и блокирование) элемента Alu было значительно выше у потомков долгожителей. Даже в старости люди, получившие в наследство от родителей хорошее здоровье, мало чем отличались на молекулярно-генетическом уровне от молодых людей. И такие потенциально опасные элементы генома, как ретротранспозоны, были у них надежно блокированы. Можно с большой долей уверенности говорить, что хорошее здоровье (как, впрочем, и плохое) передается по наследству.
Понимание природы эпигенетики показало, что родители ответственны за здоровье своих детей гораздо в большей степени, чем это считалось ранее. Совсем недавно было показано, что такая привычная сегодня вещь, как лишний вес будущих родителей, может самым негативным образом сказаться на их потомстве. Известный американский генетик Рэнди Джиртл и его коллеги из Университета Дьюка провели исследования ДНК лейкоцитов из пуповинной крови младенцев, родившихся в госпитале при их университете. По словам ученых, анализ зафиксировал существенное снижение уровня метилирования гена инсулиноподобного фактора роста 2 (IGF 2) у тех детей, чьи родители имели лишний вес: «Мы обнаружили у новорождённых, отцы которых страдали ожирением, значительное снижение метилирования IGF2 в ДНК, извлеченной из лейкоцитов пуповинной крови. Понижение уровня метилирования IGF2 связано с повышенным риском развития раковых заболеваний» [17].
Возрастные отклонения и эпигеном
Уже известно, что возрастные изменения эпигенома тесно связаны с другими возрастными явлениями, такими как увеличение продукции активных форм кислорода, укорочение теломер и т.д. Активные формы кислорода (АФК) и связанные с ними процессы с легкой руки знаменитого американского биолога Денгама Хармана (и не менее знаменитого российского биолога В.П. Скулачёва) рассматриваются сегодня некоторыми учеными в качестве одного из основных факторов старения. Хотя сами АФК участвуют во многих физиологических процессах (особенно по части иммунной системы), с возрастом может происходить опасное повышение их уровня. Обладая большой химической активностью, АФК потенциально способны повреждать мембраны клеток, митохондриальную и ядерную ДНК . Они задействованы во многих возрастных процессах и связанных с ними патологиях: атеросклерозе, болезни Альцгеймера и др. Так, к примеру, при сердечно-сосудистых болезнях АФК блокируют производство белков-сиртуинов (в частности, SIRT1), что ведет к цепи отклонений и развитию патологий (рис. 3). При болезни Альцгеймера АФК участвуют в патологических процессах образования амилоида, а также способствуют активации провоспалительных факторов — например, ядерного фактора транскрипции NF-kB [18].
Подробнее АФК и связанные с ними процессы рассмотрены в статьях «Активный кислород: друг или враг, или о пользе и вреде антиоксидантов» [19] и «Антиоксиданты против пиелонефрита» [20].

Рисунок 3. Окислительный стресс, эпигенетика и болезни сердца, легких и нервной системы. ROS — активные формы кислорода; SIRT1 — sirtuin 1, белок из семейства сиртуинов; iNOS — индуцируемая синтаза оксида азота; eNOS — эндотелиальная синтаза оксида азота; NO — оксид азота; 8-oxo — 8-гидроксигуанин, маркер окислительного повреждения ДНК; HDAC2 — гистондеацетилаза 2; NF-kB — ядерный фактор транскрипции каппа-B; p21 — белок, ингибитор циклин-зависимой киназы 1A; р16 — белок, ингибитор циклин-зависимой киназы 2A.
Еще в 1994 году Зигмунд Вейцман и его коллеги из Северо-Западного университета (Чикаго) обнаружили, что окисление свободными радикалами гуанина, обязательного «напарника» цитозина в двухцепочечной ДНК, влияет на метилирование. Окисленный продукт гуанина, 8-гидроксигуанин (8-OHdG), известный маркер окислительного повреждения ДНК, не только увеличивал частоту мутаций, но и препятствовал нормальному метилированию цитозина [21]. Оказалось, что эпигеном тесным образом связан с регулированием уровня АФК: общее возрастное уменьшение метилирования ДНК развивается параллельно с повышением уровня АФК и окислительным стрессом. И эти процессы связаны между собой. Так, возрастной окислительный стресс, вызванный повышенной продукцией АФК, достоверно снижает количество уже упоминавшихся сиртуинов, к которым сегодня приковано пристальное внимание геронтологов. Сиртуины представляют собой семейство консервативных (то есть встречающихся у всех живых организмов) белков, выполняющих одну из эпигенетических функций — деацетилирование гистонов. Многочисленные эксперименты показали благотворную роль сиртуинов в поддержании здоровья и увеличении продолжительности жизни. АФК же снижают уровень этих белков, сокращая тем самым срок жизни.
Фолатный цикл, p66Shc, эпигеном и старение
Если взять наугад любую патологию из длинного списка возрастных болезней (таких, как атеросклероз, болезнь Паркинсона, диабет или ревматоидный артрит), то совершенно точно обнаружится непосредственное участие эпигенома в их развитии. Этот вопрос сегодня хорошо изучен и уже ни у кого не вызывает сомнений. К примеру, у больных атеросклерозом обнаруживается повышенный уровень токсичного для артерий гомоцистеина. И причина этого — сбой в работе сложного биохимического процесса под названием фолатный цикл, который тесно связан с метилированием ДНК и эпигеномом.
Фолатный цикл представляет собой каскад биохимических реакций, в котором задействовано большое количество ферментов (рис. 4). Для нормального протекания фолатного цикла необходимы витамины B9 (фолиевая кислота), В6 и В12. В этом цикле происходит перенос метильных групп, которые присоединяются к гомоцистеину, а избыток гомоцистеина превращается в метионин. Метионин переходит в свою активную форму, S-аденозилметионин (SАМ), который в клетке служит основным донором метильных групп, необходимых для синтеза и метилирования ДНК, РНК, белков и фосфолипидов [22].

Рисунок 4. Схема фолатного цикла. В реакции, катализируемой ферментом MTHFR, из тетрагидрофолата и серина образуется 5,10-метилентетрагидрофолат, который затем восстанавливается до 5-метилтетрагидрофолата. На следующем этапе метильная группа от 5-метилтетрагидрофолата переносится на гомоцистеин в реакции, катализируемой В12-зависимой метилтрансферазой. В результате реметилирования гомоцистеина образуется метионин. Данную реакцию катализирует цитоплазматический фермент метионинсинтаза (MTR). Для работы фермента необходим метилкобаламин, производное витамина В12. Метионинсинтаза обеспечивает преобразование гомоцистеина в метионин посредством реакции, в которой метилкобаламин выступает в роли промежуточного переносчика метильной группы. При этом происходит окисление кобаламина, и фермент MTR переходит в неактивное состояние
В фолатном цикле может произойти сбой по двум причинам: генетическим (из-за мутаций генов ферментов фолатного цикла) или алиментарным (из-за дефицита метионина, фолиевой кислоты и других витаминов группы В). Дефицит возникает, если в рационе недостает продуктов, богатых этими веществами, или если эти вещества плохо усваиваются — как правило, на фоне вредных привычек, приема лекарств, инфекций и др.
Нарушение фолатного цикла угрожает организму сразу тремя последствиями. Первое: низкий уровень одного из производных этого цикла, 5,10-метилентетрагидрофолата, приводит к разрывам в ДНК и нарушению процессов репарации [23]. Второе: возникает дефицит основного донора метильных групп, S-аденозилметионина (SAM), без которого невозможно производить метилирование ДНК. И третье последствие: нарушается метаболизм гомоцистеина, и его уровень в крови начинает расти. А далее токсичный для клеток гомоцистеин запускает цепную реакцию со множеством патологических ответвлений.
Какой это может иметь вид? Как показали работы американских и корейских биологов — С. Ким и соавторов, — повышенная концентрация гомоцистеина в крови имеет прямую связь с дисфункцией эндотелия (тонкого слоя клеток, выстилающего поверхность сосудов) и атеросклерозом [24]. Причем активным участником в повреждающем действии гомоцистеина на клетки эндотелия является белок с «дурной репутацией», p66Shc, который обладает окислительной и проапоптозной (подстрекающей клетку к самоубийству) активностью. Подопытные мыши с нокаутированным («выключенным») геном p66shc, показывали существенное увеличение продолжительности жизни [25]. При этом такие мыши были удивительным образом устойчивы к действию окислительного стресса и развитию основных патологий, традиционно связываемых со старением: гиперхолестеринемии (повышению уровня холестерина), гипергликемии (повышению уровня глюкозы) и ишемии. И даже ударные дозы алкоголя не убивали таких мышей!
Как выяснилось, гомоцистеин в повышенных концентрациях способен увеличивать экспрессию эндотелиального p66shc посредством гипометилирования конкретных динуклеотидов CpG в промоторе (регуляторном участке гена) p66shc. И этот механизм играет важную роль в вызванной гомоцистеином эндотелиальной патологии. Дело в том, что p66Shc повышает продукцию АФК, которые признаны одной из основных причин возрастной дисфункции эндотелия [24]. Примечательно, что точно таким же действием на p66Shc, как гомоцистеин, обладают и липопротеины низкой плотности (ЛПНП, или, по-обывательски, «плохой» холестерин), рассматриваемые как один из факторов развития метаболического синдрома, которому подвержено огромное количество людей в мире. Выяснилось, что ЛПНП в состоянии вызывать гипометилирование двух динуклеотидов CpG и ацетилирование гистона 3 в промоторе p66shc, что ведет к повышению его активности [26].
Итальянский исследователь Джовамбаттиста Пани связал возрастную активность p66Shc и АФК с еще одним белком, вызывающим повышенный интерес у геронтологов, — mTOR (мишень рапамицина у млекопитающих), который регулирует в клетке процессы, связанные с развитием и клеточным ростом (рис. 5) [27]. По мнению одного из самых авторитетных исследователей mTOR, Михаила Благосклонного (Roswell Park Cancer Institute, США), этот белок занимает одно из центральных мест в процессах старения живых организмов, стимулируя развитие возрастных патологий, укорачивающих жизнь [28].

Рисунок 5. Две модели, показывающие, как p66Shc может объединять АФК и mTOR/S6K-каскад в процессе старения. а — АФК активируют белок p66Shc (или просто p66), который в свою очередь активирует рибосомную S6-киназу (S6K). АФК могут производиться в митохондриях в ответ на поступление питательных веществ (нутриентов), таким образом создавая альтернативный маршрут для детекции питательных веществ посредством S6K. б — Активированная p66 киназа S6K увеличивает образование АФК в митохондриях. В этом случае р66 может стимулироваться клеточным стрессом, белком р53 или экзогенными оксидантами. В обоих примерах воздействие р66 на старение тормозится ограничением калорий, что снижает поставку нутриентов. Активация p66 происходит в результате повышенной экспрессии (показана увеличенным значком) и фосфорилирования серина (буква «Р»). Оба изменения возникают в ответ на различные стрессы в клетках млекопитающих.
Но не только дисфункция эндотелия и сердечно-сосудистые болезни связаны с гомоцистеином и повышенной активностью p66Shc. Уже известно, что p66Shc напрямую связан с отклонением, которое сегодня чаще всего рассматривают в качестве точки отсчета ускоренного старения — с резистентностью к инсулину, когда клетки перестают взаимодействовать с этим гормоном [29]. Вместе с резистентностью к инсулину развиваются липо- и глюкозотоксичность — разрушительное воздействие избытка жирных кислот (прежде всего пальмитиновой) и глюкозы на клеточные структуры. И далее объем последствий растет как снежный ком: повышение продукции провоспалительных цитокинов, накопление опасных метаболитов (диацилглицерола, сорбитола, церамида), окислительный стресс, нарушение синтеза АТФ и дисфункция митохондрий, повреждение эндотелия и атеросклероз, гликозилирование белков, накопление амилоида, повреждение клеток, ускоренное старение. Подавляющее число долгожителей не приобретает резистентности к инсулину до самой глубокой старости. И наоборот — бόльшая часть людей, преждевременно погибших от сердечно-сосудистых патологий, имела резистентность к этому гормону.
Сегодня большое внимание геронтологов приковано к механизмам благотворного воздействия на организм ограничения калорий. Как показали исследования, урезанный по калориям рацион может продлевать жизнь. Самый яркий и часто приводимый пример такого влияния — жители японского острова Окинава, которые при суточном рационе менее 2000 ккал уверенно держат первое место в мире по числу долгожителей. Суть этого феномена складывается из многих факторов, в том числе и из эпигенетических. Обнаружилось, что ограничение калорий меняет в позитивную сторону профиль метилирования ДНК: снижается метилирование генов — подавителей опухолевого роста, что ведет к их активации, и повышается метилирование онкогенов. Кроме этого, ограничение количества потребляемой глюкозы, которая дает немалую часть калорий, в эксперименте приводило к феномену расширения лимита Хейфлика — предела клеточного деления, лежащего в основе ограниченности срока жизни клетки (рис. 6). И опять это происходило вместе с изменениями эпигенома — изменениями в метилировании ДНК и модификациями гистонов, влияющими на активность ключевых генов — р16 и hTERT [30].

Рисунок 6. Влияние ограничения потребления глюкозы на продолжительность жизни. Ограничение поступления глюкозы может влиять на эпигенетическую регуляцию как в нормальных, так и в раковых клетках. В нормальных клетках это приводит к репрессии гена р16 и активации hTERT, что позволяет расширить лимит Хейфлика. Белок р16 замедляет процесс размножения клеток, ген hTERT кодирует фермент теломеразу, способный наращивать концевые участки ДНК, сокращающиеся при делении клеток, — теломеры. В предраковых клетках противоположные эффекты на p16 и hTERT приводят к апоптозу и гибели опасных клеток.
Эпигеном и теломеры
Еще один ассоциированный с возрастом процесс — укорочение теломер (повторяющихся последовательностей ДНК, стабилизирующих концевые участки хромосом, но уменьшающихся в длине при каждом клеточном делении) — также оказался тесно связанным с эпигеномом.
Американские биологи из Института Солка и Гарвардского университета, Родерик О’Салливан и его коллеги, провели исследования, выясняющие влияние клеточного деления на структуру хроматина. Как видно из работы О’Салливана и соавторов, в стареющих клетках с сильно укороченными теломерами характер упаковки ДНК в хромосомах значительно меняется [31]. Также выяснилось, что с возрастом при каждом делении клетки вместе с укорочением теломер происходит уменьшение синтеза специальных белков-гистонов . На нуклеосомные гистоны, как швейная нить на катушку, наматывается молекула ДНК, упаковываясь таким образом в ядрах клеток. Посредством ацетилирования-деацетилирования гистонов происходит регулирование плотности упаковки ДНК. Если нужно «заглушить» гены, упаковка уплотняется, и считывающие информацию белки не могут присоединиться к регуляторным нуклеотидам. Если, наоборот, работа какого-то гена необходима, хроматин «разрыхляется», упаковка ДНК становится менее плотной и доступной для регуляторных белков.
Свойства этих белков описаны в статье «Катится, катится к ДНК гистон» [32].
Очевидно, что гистоны хроматина играют очень важную роль в нормальной работе генома. И обнаруженное американскими исследователями возрастное снижение их синтеза, связанное с делением клетки и укорочением теломер, может приводить к дестабилизации генома. По предположению О`Салливана и его коллег, хронический стрессовый сигнал генерируется за счет сокращения теломер и приводит к снижению синтеза гистонов двух видов, H3 и H4. В свою очередь, это не позволяет точно восстановить ландшафт хроматина при следующем делении, и повреждение ДНК постепенно ограничивает жизнь клетки. Даже незначительные изменения в равновесии системы «ДНК — гистоны», по мнению ученых, могут нарушить синтез ДНК, архитектуру хроматина и жизнеспособность клеток.
Но, как оказалось, это не единственно возможная взаимосвязь теломер, старения и эпигенома. Не так давно проследили еще одну потенциальную связь. В.А. Галицкий и его коллеги из Института биохимии им. А.А. Палладина (Киев) описали возможность укорочения теломер на фоне возрастной геномной нестабильности. И вот как это может выглядеть.
По мнению украинских биохимиков, микроРНК в стволовых клетках поддерживают исходный профиль эпигенетических маркеров, что и лежит в основе уникальных качеств стволовых клеток (в первую очередь, их способности к долгой жизни). Но дифференцировка — превращение стволовых клеток в специализированные — требует репрессии генов некоторых микроРНК, чтобы те не мешали активности ряда нужных генов. В результате этого происходит возрастная потеря эпигенетических маркеров, снижается уровень метилирования ДНК. А это может приводить к дерепрессии «дремлющих» в ДНК ретротранспозонов и других мобильных элементов и, как следствие, к их перемещениям и повреждению ДНК. В ответ в клетке могут запуститься системы репарации ДНК, провоцирующие несанкционированные рекомбинации в участках теломер (так называемых теломерных кэпах). И по этой причине теломеры будут терять свою длину [33]. А укорочение теломер, как мы знаем, выступает одним из признанных маркеров старения организма.
Этот процесс на примере профориентации клеток крови прекрасно проиллюстрирован (в прямом смысле) в комиксе «Кем быть? Как гемопоэтическая стволовая клетка выбирает профессию» [34]. — Ред.
Заключение. «Эпигенетический дрейф»
Насколько сильно меняется эпигеном с возрастом в зависимости от внешних факторов, наглядно показали исследования ДНК близнецов, выполненные испанскими учеными, Марио Фрага и его коллегами. Изучая монозиготных (однояйцевых) близнецов, они определили, что близнецы в возрасте трех лет идентичны не только генетически, но и эпигенетически [35]. А вот у 50-летних — по-прежнему одинаковых генетически — людей возникают существенные эпигенетические различия (рис. 7). Причем тем больше, чем внушительнее географическая дистанция между местами их проживания и, следовательно, разница в условиях проживания. Это может говорить о том, что подобные различия возникают не случайно, а зависят от условий, в которых живут люди.

Рисунок 7. Разница в метилировании ДНК близнецов в возрасте трех и 50 лет. Для того чтобы определить эпигенетические различия между близнецами, исследователи разработали оригинальную методику окраски одинаковых участков (локусов) гомологичных хромосом. В случае одинаковой экспрессии эти локусы окрашивались в желтый цвет, если были гипометилированы — в красный, а если гиперметилированы — в зеленый. Так вот, если у 3-летних близнецов хромосомы были окрашены практически полностью в желтый цвет, то у 50-летних явно доминировали зеленый и красный.
Результаты всех этих исследований позволили немецкому генетику Акселю Шумахеру разработать концепцию «эпигенетического дрейфа» — постепенного изменения с возрастом профиля метилирования ДНК [36]. Согласно концепции, возрастзависимый «эпигенетический дрейф» является естественным процессом, наблюдающимся у всех, даже полностью здоровых людей. Но у тех, кто подвергается сильному влиянию окружающей среды, вредных привычек, стрессов, неправильного питания, дрейф может «давать крен» в сторону неблагоприятных профилей, негативно меняя работу генома.
О природе различных, не только эпигенетических, возрастных изменений в клетках и возможных путях их преодоления образно рассказывает статья «Зачем клетки стареют» [37]. — Ред.
После всех проведенных исследований ученые могут уверенно говорить, что основу долголетия и хорошего здоровья вместе с другими факторами во многом определяет именно состояние эпигенома. А оно складывается из двух составляющих: из того, что мы получили в наследство от своих родителей, и из привычек, которые мы выработали в нашей жизни. Как сказал один из основоположников эпигенетики, профессор МГУ Б.Ф. Ванюшин, «нет никакого сомнения в том, что метилирование ДНК и модификации гистонов, а также избирательный сайленсинг генов малыми РНК играют очень важную роль в жизни клетки и организма, поэтому дальнейшие исчерпывающие исследования в этой захватывающей области знаний — очень важная и плодотворная задача нашего века» [38]. И с этим трудно поспорить. Остается ждать новых открытий, которые подарит нам область знаний, стремительно ворвавшаяся в нашу жизнь, — наука ЭПИГЕНЕТИКА.
- Развитие и эпигенетика, или История о Минотавре;
- Эпигенетические часы: сколько лет вашему метилому?;
- Эпигенетика поведения: как бабушкин опыт отражается на ваших генах?;
- Обо всех РНК на свете, больших и малых;
- Как избавиться от РНК за несколько минут;
- МикроРНК — чем дальше в лес, тем больше дров;
- Callaway E. (2014). Epigenomics starts to make its mark. Nature. 508, 22;
- Вайсерман А.М., Войтенко В.П., Мехова Л.В. (2011). Эпигенетическая эпидемиология ассоциированных с возрастом заболеваний. Онтогенез. 42, 1–21;
- Hancks D.C. and Kazazian H.H. (2012). Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev. 22, 191–203;
- Тайны «молекулярных паразитов», или Как путешествовать по геному;
- Разнообразия много не бывает: чем занимаются мобильные элементы генома в мозге;
- Геном человека: полезная книга, или глянцевый журнал?;
- Kucharski R., Maleszka J., Foret S., Maleszka R. (2008). Nutritional control of reproductive status in honeybees via DNA methylation. Science. 319, 1827–1830;
- Lyko F., Foret S., Kucharski R., Wolf S., Falckenhayn C., Maleszka R. (2010). The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8, e1000506;
- Corona M., Velarde R.A., Remolina S., Moran-Lauter A., Wang Y., Hughes K.A., Robinson G.E. (2007). Vitellogenin, juvenile hormone, insulin signaling, and queen honey bee longevity. Proc. Natl. Acad. Sci. USA. 104, 7128–7133;
- Gentilini D., Mari D., Castaldi D., Remondini D., Ogliari G., Ostan R. et al. (2013). Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age (Dordr). 35, 1961–1973;
- Soubry A., Schildkraut J.M., Murtha A., Wang F., Huang Z., Bernal A. et al. (2013). Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 11, 29;
- Cencioni C., Spallotta F., Martelli F., Valente S., Mai A., Zeiher A.M., Gaetano C. (2013). Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 14, 17643–17663;
- Активный кислород: друг или враг, или О пользе и вреде антиоксидантов;
- Антиоксиданты против пиелонефрита;
- Weitzman S.A., Turk P.W., Milkowski D.H., Kozlowski K. (1994). Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. USA. 91, 1261–1264;
- Гречанина Е.Я., Лесовой В.Н., Мясоедов В.В., Гречанина Ю.Б., Гусар В.А. (2010). Закономерная связь между развитием некоторых эпигенетических заболеваний и нарушением метилирования ДНК вследствие дефицита ферментов фолатного цикла. Ультразвуковая перинатальная диагностика. 29, 27–59;
- Zijno A., Andreoli C., Leopardi P., Marcon F., Rossi S., Caiola S. et al. (2003). Folate status, metabolic genotype, and biomarkers of genotoxicity in healthy subjects. Carcinogenesis. 24, 1097–1103;
- Kim C.S., Kim Y.R., Naqvi A., Kumar S., Hoffman T.A., Jung S.B. et al. (2011). Homocysteine promotes human endothelial cell dysfunction via site-specific epigenetic regulation of p66shc. Cardiovasc. Res. 92, 466–475;
- Галимов Е.Р. (2010). Роль p66shc в окислительном стрессе и апоптозе. Acta Naturae. 4, 49–56;
- Kim Y.R., Kim C.S., Naqvi A., Kumar A., Kumar S., Hoffman T.A., Irani K. (2012). Epigenetic upregulation of p66shc mediates low-density lipoprotein cholesterol-induced endothelial cell dysfunction. Am. J. Physiol. Heart Circ. Physiol. 303, 189–196;
- Pani G. (2010). P66SHC and ageing: ROS and TOR? Aging (Albany NY). 2, 514–518;
- Blagosklonny M.V. (2013). MTOR-driven quasi-programmed aging as a disposable soma theory: blind watchmaker vs. intelligent designer. Cell Cycle. 12, 1842–1847;
- Ranieri S.C., Fusco S., Panieri E., Labate V., Mele M., Tesor V., Ferrara A.M. et al. (2010). Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA. 107, 13420–13428;
- Tollefsbol T. (2014). Dietary epigenetics in cancer and aging. Cancer Treat. Res. 159, 10;
- O’Sullivan R.J., Kubicek S., Schreiber S.L., Karlseder J. (2010). Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 17, 1218–1225;
- Катится, катится к ДНК гистон;
- Галицкий В.А. (2009). Эпигенетическая природа старения. Цитология. 5, 388–397;
- Кем быть? Как гемопоэтическая стволовая клетка выбирает профессию;
- Fraga M.F., Ballestar E., Paz M.F., Ropero S., Setien F., Ballestar M.L. et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA. 102, 10604–10609;
- Wang S.C., Oelze B., Schumacher A. (2008). Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE. 3, e2698;
- Зачем клетки стареют;
- Ванюшин Б.Ф. (2013). Эпигенетика сегодня и завтра. Вавиловский журнал генетики и селекции. 17, 805–832.
- Авторы
- Файлы
- Ключевые слова
- Литература
Зобкова Н.В.
1
Аншакова М.М.
1
1 ФГБОУ ВО «Оренбургский медицинский университет»
старение
теория
биомаркеры
1.[https://www.researchgate.net/publication/329101904_GENETICESKIE_I_EPIGENETICESKIE_MEHANIZMY_STARENIA]
2. [ https://testdnk.pro/informacia/metilirovanie-dnk.html]
3. [ https://dolgo-jv.ru/inflamaging.html]
4. [ http://pro-starenie.ru/glikirovanie-belkov-starenie-i-omolozhenie-organizma]
5.[ https://dolgo-jv.ru/glycation_and_aging.html]
Старение является сложным процессом. Физиологические изменения, которые происходят в теле человека с возрастом, в первую очередь выражаются в снижении биологических функций и способности приспосабливаться к метаболическому стрессу. Эти физиологические изменения обычно сопровождаются психологическими и поведенческими изменениями. Как с теоретической, так и с практической точки зрения также предполагается, что вряд ли когда-либо будет найдена какая-либо единственная биологическая мера, которая могла бы точно оценить скорость старения сложного организма, такого как человек. Таким образом, предполагается, что для оценки скорости старения такого организма необходимо будет создать панели биомаркеров, состоящие из множества показателей. Эти панели биомаркеров, вероятно, будут состоять из показателей, которые оценивают все или многие системы органов человека, такие как сердечно-сосудистая система, мозг, печень, кости, память и так далее.
Биомаркеры старения — это измеряемые показатели жизнедеятельности, которые воспроизводимо изменяются, количественно и качественно, с возрастом организма. Они могут иметь место на различных уровнях организации живой системы: они могут быть системными, например изменения в иммунной системе, в системе крови, в нейропсихических функциях, в функции почек; могут быть на клеточном уровне, например так называемое клеточное старение, когда в норме делящиеся клетки отказываются делиться, переходят в состояние покоя и уже не возвращаются в деление; могут быть на молекулярном уровне, например поломки хромосом или так называемая генетическая нестабильность, когда с возрастом ДНК повреждается, эти повреждения в клетках накапливаются, и их можно детектировать и определять возраст тканей или конкретных клеток по этим показателям.
Рассмотрим процессы старения с биохимической точки зрения: процессы гликирования, воспаления, метилирования и свободные радикалы.
Гликирование, или гликозилирование, обусловлено способностью глюкозы образовывать с аминогруппами различных белков, возможно и с ДНК, различные соединения (интермедиаты), участвующие в обмене и являющиеся исходным материалом для образования необратимых в химических реакциях веществ, которые получили название конечных продуктов гликозилирования (КПГ). Период полураспада этих продуктов более длительный, чем белков (от нескольких месяцев до нескольких лет). Скорость образования КПГ зависит от уровня и длительности экспозиции глюкозы. Накапливание гликированных белков, как и продуктов окисления, которое происходит со временем, приводит к возрастным изменениям.
Гликирование белков — один с основных факторов, порождающих изнашивание организма, вызывающих склероз, кровоизлияние в мозг, разрыв сердца, морщины и т.п. В нормальных условиях темп гликирования белков в организме до такой степени незначителен, что продукты успевают удаляться. Особенно это выражено в крови, где резко повышается уровень поврежденных белков.
Рис.1 Гликирование ДНК
В основе эпигенетической теории старения лежит процесс метилирования.
Теория принадлежит к группе теорий произвольных повреждений клеток и утверждает, что старение организма имеет непосредственную зависимость от накоплений повреждений в клетках, наносимых свободными радикалами (свободные радикалы – это все молекулы и атомы, содержащие один или более неспаренных электронов на внешнем электронном уровне) в течение жизни.
Теория предполагает, что эрозия теломер начинает ускоряться в десятки или даже в сотни раз из-за рекомбинаций в их ДНК, которые вызваны функционированием клеточных систем репарации ДНК.
Метилирование ДНК представляет один из эпигенетических механизмов регуляции экспрессии генов. В ходе метилирования метильная группа СН3- специальными ферментами присоединяется к одному из оснований ДНК, а именно к цитозину. В результате образуется 5-метилцитозин и происходит инактивация экспрессии генов — процесс транскрипции блокируется. Как известно на сегодняшний день, метилирование ДНК – это процесс динамический. Оно может изменяться под воздействием каких-либо внешних факторов, связано с развитием ряда патологий и может наследоваться несколькими следующими поколениями. Метилирование играет одну из важнейших ролей в деактивации чужеродной ДНК, а также и в таких процессах как развития и старения. Стоит заметить, что с возрастом наблюдается гипометилирование (деметилирование) и связанная с этим хромосомная нестабильность. Помимо этого, при старении происходит и обратный процесс – гиперметилирование некоторых промоторных областей, в том числе определенных генов-супрессоров опухолей, что связано с развитием патологий.
Рис.2 Метилирование ДНК
Воспаление старения представляет одну из ключевых ролей в появлении и формировании возрастных заболеваний (например, инсулиннезависимый диабет, болезнь Альцгеймера, сердечно-сосудистые заболевания, слабость, остеопороз и рак).
Воспаление считается составляющей сложного биологического ответа тканей организма на вредоносные раздражители, также предполагает защитный ответ организма.
Роль воспаления заключается в ликвидации изначальной первопричины дефекта клеток, очищении клеток и тканей от воспалительного процесса, а также восстановлении тканей.
Старение характеризуется системным хроническим воспалением. Воспаление можно определить с помощью маркеров — С-реактивного белка и интерлейкина-6.
Согласно данной теории, одной из главных причин появления признаков старости являются активные формы кислорода (АФК), которые являются побочным продуктом реакции окисления. Получившиеся АФК вступают в реакцию с молекулами клеток, повреждая их. А это приводит к апоптозу клетки.
В принципе все это нормально и наш организм постоянно проводит обновление (например, ранки на коже, видимая часть). Однако часть тканей у нас почти не обновляется (хрящи, сухожилия, кристаллин в хрусталике) и там апоптоз постепенно приводит к проявлению всем знакомым признакам старения.
Также вторая часть проблемы повышенного уровня АФК, в том, что повышается вероятность повреждения ДНК и перерождения клетки в раковую. А если это происходит, то все становится плохо.
С возрастом чрезмерное накопление свободных радикалов приводит к накапливанию ошибок в ДНК и ведет к запусканию процесса старения человека и что эти повреждения происходят по вине чрезмерного накопления свободных радикалов. Если число дефектов ДНК достигают предельного степени (с возрастом), в таком случае начинается старение — появляются злокачественные опухоли, также прочие заболевания старости. В природе существуют вещества, которые способны нейтрализовать свободные радикалы. Это антиоксиданты. Большое количество антиоксидантов содержатся в овощах и фруктах
Таким образом, старение является результатом многих биохимических процессов. Каждый фактор, участвующий в возрастных изменениях организма, тем или иным образом влияет на его функционирование.
Библиографическая ссылка
Зобкова Н.В., Аншакова М.М. БИОМАРКЕРЫ ПРОЦЕССА СТАРЕНИЯ // Международный студенческий научный вестник. – 2021. – № 2.
;
URL: https://eduherald.ru/ru/article/view?id=20613 (дата обращения: 13.06.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)