
Механизмы исправления ошибок во время репликации ДНК и ее репарация вследствие повреждений на протяжении всего жизненного цикла клетки.
Основные моменты:
-
Клетки имеют различные механизмы предотвращения возникновения мутаций – необратимых изменений в ДНК
-
В процессе синтеза ДНК, большинство ДНК-полимераз «проверяют свою работу» и проводят замену бо́льшей части ошибочно вставленных нуклеотидов. Этот процесс можно назвать исправлением ошибок.
-
Сразу после синтеза ДНК любые оставшиеся ошибочные нуклеотиды обнаруживаются и заменяются в так называемом процессе репарации ошибочно спаренных нуклеотидов.
-
Если ДНК повреждена, она может быть восстановлена с помощью различных механизмов, например, путём прямой репарации, эксцизионной репарации или путём восстановления двухцепочечных разрывов
- пострепликативной репарации.
Введение
Как ДНК связана с раком? Рак возникает при неконтролируемом делении клеток, когда игнорируются клеточные «стоп»-сигналы, что приводит к образованию опухоли. Это неправильное поведение клеток вызвано накопившимися мутациями — необратимыми изменениями последовательности ДНК клетки.
На самом деле, ошибки в процессе репликации и повреждения ДНК возникают в клетках нашего тела постоянно. Однако в большинстве случаев они не приводят к раку и даже не вызывают мутаций, такие ошибки обычно обнаруживаются и исправляются в процессе репарации ДНК. Если же повреждение исправить не удаётся, то в клетке включается механизм самоуничтожения — (апоптоз), который предотвращает передачу поврежденной ДНК дочерним клеткам.
Мутации возникают и передаются дочерним клеткам только тогда, когда эти механизмы не справляются. В частности, рак возникает в случае накопившихся в одной клетке мутаций генов, связанных с делением.
В этой статье мы подробно рассмотрим механизмы, используемые клетками для исправления ошибок, которые возникают в процессе репликации. К ним относятся:
-
Исправление ошибок – процесс, который возникает во время репликации ДНК.
-
Репарация ошибочно спаренных нуклеотидов, которая происходит сразу же после репликации ДНК.
-
Механизмы репарации, которые выявляют и исправляют повреждения ДНК на протяжении всего клеточного цикла
Исправление ошибок
ДНК-полимеразы — это ферменты, участвующие в репликации ДНК. Во время копирования ДНК большинство ДНК-полимераз «проверяют», корректный ли нуклеотид они добавляют. Этот процесс называется исправлением ошибок. Если полимераза обнаружит, что был добавлен неправильный нуклеотид, она сразу же удалит и заменит его и только после этого продолжит синтез ДНКstart superscript, 1, end superscript.
Репарация ошибочно спаренных нуклеотидов
Процесс исправления избавляет от основной массы ошибок, но не от всех. После создания новой ДНК запускается механизм репарации ошибочно спаренных нуклеотидов — удаления и замены ошибочно спаренных нуклеотидов, оставшихся в результате репликации. Исправление несоответствий между парами оснований также может включать в себя исправление небольших вставок и делеций, возникающих вследствие «соскальзывания» полимеразы с исходной цепи squared.
Как происходит восстановление неправильно спаренных нуклеотидов? Во-первых, белковый комплекс распознаёт неправильно спаренный нуклеотид и связывается с ним. Другой комплекс разрезает ДНК в области несовпадения, а ещё одна группа ферментов отщепляет некорректный нуклеотид вместе с небольшим участком вокруг него. Затем ДНК-полимераза заполняет этот пробел правильными нуклеотидами, а фермент ДНК-лигаза сшивает разрывы в цепиsquared.
Удивительно: как белки, участвующие в восстановлении ДНК, определяют, «кто прав» во время репарации ошибочно спаренных нуклеотидов? То есть, когда два основания неправильно соединены (как G (гуанин) и T (тимин) на рисунке выше), какое из этих двух оснований должно быть удалено и заменено?
У бактерий можно отличить исходную и дочернюю цепи ДНК по метилированным основаниям. На исходной цепи ДНК есть метильные (minus, start text, C, H, end text, start subscript, 3, end subscript) группы, присоединенные к некоторым из ее оснований, а у дочерней цепи таких групп еще нетcubed.
У эукариот процессы, позволяющие идентифицировать исходную цепь при устранении несоответствий, включают распознавание одноцепочечных разрывов, которые обнаруживаются только у дочерней цепи cubed.
Механизмы репарации ДНК
С ДНК может что-нибудь случиться практически в любой момент жизни клетки, а не только во время репликации. Фактически, ДНК постоянно повреждается из-за воздействия внешних факторов: ультрафиолетового излучения и радиации, химических веществ, не говоря уже о спонтанных процессах, которые протекают даже без вмешательства окружающей среды!start superscript, 4, end superscript
К счастью, наши клетки имеют механизмы восстановления, с помощью которых они находят и исправляют большинство повреждений ДНК. Можно выделить несколько типов репарации:
-
Прямая репарация. Некоторые повреждения ДНК, вызванные химическими реакциями, могут быть «исправлены» находящимися в клетке ферментами.
-
Эксцизионная репарация. Повреждение одного или нескольких нуклеотидов ДНК часто исправляется удалением и заменой поврежденного участка. При эксцизионной репарации оснований удаляется только поврежденное основание. В случае эксцизионной репарации нуклеотидов, как и в случае репарации ошибочно спаренных нуклеотидов, которое мы рассмотрели выше, удаляются целиком нуклеотиды.
-
Репарация двухцепочечных разрывов: Существуют два основных способа: негомологичное соединение концов и гомологичная рекомбинация. Они используются для восстановления двухцепочечных разрывов ДНК (когда вся хромосома разделяется на две части).
Прямая репарация
В некоторых случаях клетка может исправить повреждение ДНК, обратив вызвавшую его реакцию. Дело в том, что «повреждение ДНК» — это, как правило, присоединение к ней лишней группы в результате химической реакции.
Например, гуанин (G) может подвергаться реакции с присоединением метильной (minus, start text, C, H, end text, start subscript, 3, end subscript) группы к атому кислорода в азотистом основании. Если это не исправить, метил-содержащий гуанин будет связываться с тимином (Т), а не с цитозином (С) во время репликации ДНК. К счастью, у людей и многих других организмов есть фермент, который может удалить метильную группу, обратив реакцию, и тем самым вернуть азотистое основание в нормальное состояниеstart superscript, 5, end superscript.
Эксцизионная репарация оснований
Эксцизионная репарация оснований — это механизм, используемый для обнаружения и удаления определенных типов поврежденных азотистых оснований. Ключевую роль в нем играет группа ферментов, называемых гликозилазами. Каждая гликозилаза обнаруживает и удаляет определенный вид поврежденных оснований.
Например, в процессе реакции дезаминирования цитозин может превратиться в урацил — основание, обычно встречающееся только в РНК. Во время репликации ДНК урацил будет соединяться с аденином, а не с гуанином (в отличие от цитозина), поэтому такое превращение может привести к возникновению мутацииstart superscript, 5, end superscript.
Для предотвращения подобных изменений гликозилаза, являющаяся частью сигнального пути эксцизионной репарации, обнаруживает и удаляет дезаминированные цитозины. После того, как основание было удалено, удаляется и оставшаяся часть нуклеотида, а другие ферменты заполняют пробелstart superscript, 6, end superscript.
Эксцизионная репарация нуклеотидов
Эксцизионная репарация нуклеотидов — это еще один способ удаления и замены поврежденных оснований. В результате нее обнаруживаются и корректируются повреждения, которые искажают форму двойной спирали ДНК. Например, азотистые основания могут измениться, присоединив к себе громоздкие группы атомов, в частности, в результате воздействия химических веществ, содержащихся в сигаретном дымеstart superscript, 7, end superscript.
Эксцизионная репарация нуклеотидов также используется для устранения повреждений, вызванных ультрафиолетовым излучением, например, при получении солнечного ожога. Под воздействием УФ-излучения цитозин и тимин могут вступать в реакцию с соседними основаниями, которые также являются цитозином или тимином, образуя при этом связи, изменяющие форму двойной спирали и вызывающие ошибки в процессе репликации ДНК. Наиболее распространенный тип таких связей — тиминовый димер — он состоит из двух тиминовых оснований, вступающих в реакцию друг с другом и образующих химическую связьstart superscript, 8, end superscript.
При эксцизионной репарации нуклеотидов поврежденные нуклеотиды удаляются вместе с соседними нуклеотидами. В этом процессе хеликаза (фермент, раскручивающий ДНК) раскрывает ДНК, образуя пузырь, а ферменты, разрезающие ДНК, отсекают поврежденную часть пузыря. Полимераза заполняет пробел, а лигаза сшивает разрыв в цепиstart superscript, 9, end superscript.
Репарация двухцепочечных разрывов
Некоторые факторы окружающей среды, например, радиация, могут вызывать разрывы обеих цепочек ДНК (разделение хромосомы на две части). Такие повреждения ДНК, если верить комиксам, ведут к появлению супергероев, но могут встречаться и после реальных катастроф, например, Чернобыльской.
Двухцепочечные разрывы опасны, потому что большие сегменты хромосом и сотни содержащихся в них генов могут быть потеряны, если разрыв не будет восстановлен. Существует два способа восстановления двухцепочечных разрывов ДНК: негомологичное соединение концов и гомологичная рекомбинация.
При негомологичном соединении концов два разорванных конца хромосомы просто склеиваются обратно. Этот механизм восстановления является «грубым» и неточным, в результате в месте разрыва, как правило, либо теряются нуклеотиды, либо добавляются лишние, что может привести к мутациям. Но это в любом случае лучше потери целого фрагмента хромосомыstart superscript, 10, end superscript.
При гомологичной рекомбинации для восстановления разрыва используется фрагмент из гомологичной хромосомы, который соответствует поврежденной хромосоме (или из сестринской хроматиды, если ДНК была реплицирована). В этом процессе две хромосомы объединяются, и неповрежденная область гомологичной хромосомы или хроматиды используется в качестве матрицы для замены поврежденной области. Гомологичная рекомбинация работает «чище», точнее, чем негомологичное соединение концов, и обычно не приводит к образованию мутацийstart superscript, 11, end superscript.
Репарация ДНК и заболевания человека
Доказательства важности механизмов репарации получены на основе генетических заболеваний человека. Во многих случаях мутации в генах, которые кодируют белки, участвующие в репарации, связаны с наследственным раком. Например:
-
Наследственный неполипозный колоректальный рак (также называемый синдромом Линча) вызван мутациями в генах, кодирующих белки, которые участвуют в репарации ошибочно спаренных нуклеотидовstart superscript, 12, comma, 13, end superscript. Поскольку такие нуклеотиды не восстанавливаются, у людей, страдающих этим синдромом, мутации накапливаются гораздо быстрее, чем у здоровых. Это может привести к развитию опухолей толстой кишки.
-
Люди с пигментной ксеродермой очень чувствительны к ультрафиолетовому излучению. Это вызвано мутациями в белках, участвующих в эксцизионной репарации нуклеотидов. Когда они не функционируют, димеры тимина и другие виды повреждений, вызванные ультрафиолетовым излучением, перестают восстанавливаться. У людей с пигментной ксеродермой после нескольких минут пребывания на солнце могут возникнуть сильные солнечные ожоги, и около половины из них заболевают раком кожи в возрасте до 10 лет, если только они не избегают солнечных лучейstart superscript, 14, end superscript.
Очевидно, что частые шибки при
воспроизведении генетической информации
в процессе репликации, могут подвергнуть
большому риску сохранность видов и их
жизнеспособность. Было установлено,
что частота ошибок при репликации не
превышает 1 ошибки на 109–1010
нуклеотидов. В то же время комплементарность
оснований может обеспечить лишь
существенно меньшую верность
воспроизведения — 1 ошибку на 104–105
оснований. Какие же дополнительные
механизмы повышают верность репликации
еще сто тысяч раз?
Выше уже было сказано, что ДНК-полимеразы
I и III кроме
полимеразной активности обладают еще
и 3-экзонуклеазной
активностью. Оказалось, что если
ДНК-полимераза встраивает неправильный
нуклеотид, она делает шаг назад, отщепляет
этот нуклеотид и повторно включает в
растущую цепь уже правильный нуклеотид.
Возможно, что существуют, особенно в
эукариотических клетках, и другие
механизмы исправления ошибок, возникающих
в процессе репликации.
Интересно, что некоторые эукариотические
ДНК-полимеразы не осуществляют такую
корректировку. По-видимому, в этом случае
точность процесса репликации обеспечивается
с помощью каких-то других средств.
1.2.Мутагенез
Молекулы ДНК живых организмов неизбежно
подвергаются действию различных
повреждающих факторов: химических
реагентов, ультрафиолетового излучения,
и более жесткой радиации (фонового
радиоактивного излучения горных пород,
космических лучей и техногенной
радиации). При этом возникают повреждения
в ДНК. Значительная часть таких повреждений
ДНК исправляется сразу по их возникновении
(см. Раздел 1.2.1,»). Неисправленные
повреждения, передающиеся по наследству,
называются мутациями.
Мутации могут быть нескольких видов.
Изменение одной пары оснований называют
точечной мутацией (замена одного
единственного основания называется
мутацией замещения). Подобная мутация
вызывает замену одной аминокислоты в
полипептиде, кодируемом данным геном.
Если такая замена происходит в вариабельной
части белка, то она мало или совсем не
сказывается на жизнедеятельности
клетки. Если же «неправильная» аминокислота
оказывается в активном центре фермента,
то это, как правило, приводит к потере
ферментом каталитической активности
и к гибели клетки. В редких случаях
полипептидный продукт, получаемый их
мутантного гена, оказывается лучше
приспособленным к выполнению своей
функции в тех новых условиях, в которые
попал организм. Такие мутации дают
потомству преимущества в борьбе за
существование, и серия соответствующих
мутаций может привести к появлению
нового вида.
Точечные мутации замещения составляют
лишь небольшую часть мутаций. Более
многочисленными и более опасными для
клеток мутациями являются мутации,
связанные с вставками и делециями
(вырезанием) нуклеотидов.
1.2.1.Репарация днк
Для исправления повреждений, возникающих
в одной из цепей ДНК, в клетке существует
большая группа ферментов репарации.
Наиболее распространенную стратегию
репарации мы рассмотрим на примере
исправления повреждений, возникающих
при действии такого сильного мутагена,
как азотистая кислота. Основным
результатом действия азотистой кислоты
является превращение цитозина в урацил.
В клетке есть специальный фермент,
урацил-ДНК-гликозидаза, который опознает
урацил и гидролизует гликозидную связь
между урацилом и остатком дезоксирибозы.
В результате в цепи ДНК появляется
дезоксирибозный остаток, не несущий
никакого основания. Появление такого
остатка служит сигналом специальной
эндонуклеазе к выщеплению этого остатка
из цепи ДНК. Образующаяся брешь
застраивается ДНК-полимеразой (специальный
фермент, работающий в системе репарации)
по информации, содержащейся в
противоположной цепи ДНК.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
From Wikipedia, the free encyclopedia
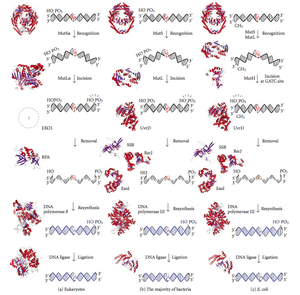
Diagram of DNA mismatch repair pathways. The first column depicts mismatch repair in eukaryotes, while the second depicts repair in most bacteria. The third column shows mismatch repair, to be specific in E. coli.
DNA mismatch repair (MMR) is a system for recognizing and repairing erroneous insertion, deletion, and mis-incorporation of bases that can arise during DNA replication and recombination, as well as repairing some forms of DNA damage.[1][2]
Mismatch repair is strand-specific. During DNA synthesis the newly synthesised (daughter) strand will commonly include errors. In order to begin repair, the mismatch repair machinery distinguishes the newly synthesised strand from the template (parental). In gram-negative bacteria, transient hemimethylation distinguishes the strands (the parental is methylated and daughter is not). However, in other prokaryotes and eukaryotes, the exact mechanism is not clear. It is suspected that, in eukaryotes, newly synthesized lagging-strand DNA transiently contains nicks (before being sealed by DNA ligase) and provides a signal that directs mismatch proofreading systems to the appropriate strand. This implies that these nicks must be present in the leading strand, and evidence for this has recently been found.[3]
Recent work[4] has shown that nicks are sites for RFC-dependent loading of the replication sliding clamp, proliferating cell nuclear antigen (PCNA), in an orientation-specific manner, such that one face of the donut-shape protein is juxtaposed toward the 3′-OH end at the nick. Loaded PCNA then directs the action of the MutLalpha endonuclease [5] to the daughter strand in the presence of a mismatch and MutSalpha or MutSbeta.
Any mutational event that disrupts the superhelical structure of DNA carries with it the potential to compromise the genetic stability of a cell. The fact that the damage detection and repair systems are as complex as the replication machinery itself highlights the importance evolution has attached to DNA fidelity.
Examples of mismatched bases include a G/T or A/C pairing (see DNA repair). Mismatches are commonly due to tautomerization of bases during DNA replication. The damage is repaired by recognition of the deformity caused by the mismatch, determining the template and non-template strand, and excising the wrongly incorporated base and replacing it with the correct nucleotide. The removal process involves more than just the mismatched nucleotide itself. A few or up to thousands of base pairs of the newly synthesized DNA strand can be removed.
Mismatch repair proteins[edit]
| DNA mismatch repair protein, C-terminal domain | |||||||
|---|---|---|---|---|---|---|---|

hpms2-atpgs |
|||||||
| Identifiers | |||||||
| Symbol | DNA_mis_repair | ||||||
| Pfam | PF01119 | ||||||
| Pfam clan | CL0329 | ||||||
| InterPro | IPR013507 | ||||||
| PROSITE | PDOC00057 | ||||||
| SCOP2 | 1bkn / SCOPe / SUPFAM | ||||||
|
Mismatch repair is a highly conserved process from prokaryotes to eukaryotes. The first evidence for mismatch repair was obtained from S. pneumoniae (the hexA and hexB genes). Subsequent work on E. coli has identified a number of genes that, when mutationally inactivated, cause hypermutable strains. The gene products are, therefore, called the «Mut» proteins, and are the major active components of the mismatch repair system. Three of these proteins are essential in detecting the mismatch and directing repair machinery to it: MutS, MutH and MutL (MutS is a homologue of HexA and MutL of HexB).
MutS forms a dimer (MutS2) that recognises the mismatched base on the daughter strand and binds the mutated DNA. MutH binds at hemimethylated sites along the daughter DNA, but its action is latent, being activated only upon contact by a MutL dimer (MutL2), which binds the MutS-DNA complex and acts as a mediator between MutS2 and MutH, activating the latter. The DNA is looped out to search for the nearest d(GATC) methylation site to the mismatch, which could be up to 1 kb away. Upon activation by the MutS-DNA complex, MutH nicks the daughter strand near the hemimethylated site. MutL recruits UvrD helicase (DNA Helicase II) to separate the two strands with a specific 3′ to 5′ polarity. The entire MutSHL complex then slides along the DNA in the direction of the mismatch, liberating the strand to be excised as it goes. An exonuclease trails the complex and digests the ss-DNA tail. The exonuclease recruited is dependent on which side of the mismatch MutH incises the strand – 5′ or 3′. If the nick made by MutH is on the 5′ end of the mismatch, either RecJ or ExoVII (both 5′ to 3′ exonucleases) is used. If, however, the nick is on the 3′ end of the mismatch, ExoI (a 3′ to 5′ enzyme) is used.
The entire process ends past the mismatch site — i.e., both the site itself and its surrounding nucleotides are fully excised. The single-strand gap created by the exonuclease can then be repaired by DNA Polymerase III (assisted by single-strand-binding protein), which uses the other strand as a template, and finally sealed by DNA ligase. DNA methylase then rapidly methylates the daughter strand.
MutS homologs[edit]
When bound, the MutS2 dimer bends the DNA helix and shields approximately 20 base pairs. It has weak ATPase activity, and binding of ATP leads to the formation of tertiary structures on the surface of the molecule. The crystal structure of MutS reveals that it is exceptionally asymmetric, and, while its active conformation is a dimer, only one of the two halves interacts with the mismatch site.
In eukaryotes, MutS homologs form two major heterodimers: Msh2/Msh6 (MutSα) and Msh2/Msh3 (MutSβ). The MutSα pathway is involved primarily in base substitution and small-loop mismatch repair. The MutSβ pathway is also involved in small-loop repair, in addition to large-loop (~10 nucleotide loops) repair. However, MutSβ does not repair base substitutions.
MutL homologs[edit]
MutL also has weak ATPase activity (it uses ATP for purposes of movement). It forms a complex with MutS and MutH, increasing the MutS footprint on the DNA.
However, the processivity (the distance the enzyme can move along the DNA before dissociating) of UvrD is only ~40–50 bp. Because the distance between the nick created by MutH and the mismatch can average ~600 bp, if there is not another UvrD loaded the unwound section is then free to re-anneal to its complementary strand, forcing the process to start over. However, when assisted by MutL, the rate of UvrD loading is greatly increased. While the processivity (and ATP utilisation) of the individual UvrD molecules remains the same, the total effect on the DNA is boosted considerably; the DNA has no chance to re-anneal, as each UvrD unwinds 40-50 bp of DNA, dissociates, and then is immediately replaced by another UvrD, repeating the process. This exposes large sections of DNA to exonuclease digestion, allowing for quick excision (and later replacement) of the incorrect DNA.
Eukaryotes have five MutL homologs designated as MLH1, MLH2, MLH3, PMS1, and PMS2. They form heterodimers that mimic MutL in E. coli. The human homologs of prokaryotic MutL form three complexes referred to as MutLα, MutLβ, and MutLγ. The MutLα complex is made of MLH1 and PMS2 subunits, the MutLβ heterodimer is made of MLH1 and PMS1, whereas MutLγ is made of MLH1 and MLH3. MutLα acts as an endonuclease that introduces strand breaks in the daughter strand upon activation by mismatch and other required proteins, MutSα and PCNA. These strand interruptions serve as entry points for an exonuclease activity that removes mismatched DNA. Roles played by MutLβ and MutLγ in mismatch repair are less-understood.
MutH: an endonuclease present in E. coli and Salmonella[edit]
MutH is a very weak endonuclease that is activated once bound to MutL (which itself is bound to MutS). It nicks unmethylated DNA and the unmethylated strand of hemimethylated DNA but does not nick fully methylated DNA. Experiments have shown that mismatch repair is random if neither strand is methylated.[citation needed] These behaviours led to the proposal that MutH determines which strand contains the mismatch.
MutH has no eukaryotic homolog. Its endonuclease function is taken up by MutL homologs, which have some specialized 5′-3′ exonuclease activity. The strand bias for removing mismatches from the newly synthesized daughter strand in eukaryotes may be provided by the free 3′ ends of Okazaki fragments in the new strand created during replication.
PCNA β-sliding clamp[edit]
PCNA and the β-sliding clamp associate with MutSα/β and MutS, respectively. Although initial reports suggested that the PCNA-MutSα complex may enhance mismatch recognition,[6] it has been recently demonstrated[7] that there is no apparent change in affinity of MutSα for a mismatch in the presence or absence of PCNA. Furthermore, mutants of MutSα that are unable to interact with PCNA in vitro exhibit the capacity to carry out mismatch recognition and mismatch excision to near wild type levels. Such mutants are defective in the repair reaction directed by a 5′ strand break, suggesting for the first time MutSα function in a post-excision step of the reaction.
Clinical significance[edit]
Inherited defects in mismatch repair[edit]
Mutations in the human homologues of the Mut proteins affect genomic stability, which can result in microsatellite instability (MSI), implicated in some human cancers. In specific, the hereditary nonpolyposis colorectal cancers (HNPCC or Lynch syndrome) are attributed to damaging germline variants in the genes encoding the MutS and MutL homologues MSH2 and MLH1 respectively, which are thus classified as tumour suppressor genes. One subtype of HNPCC, the Muir-Torre Syndrome (MTS), is associated with skin tumors. If both inherited copies (alleles) of a MMR gene bear damaging genetic variants, this results in a very rare and severe condition: the mismatch repair cancer syndrome (or constitutional mismatch repair deficiency, CMMR-D), manifesting as multiple occurrences of tumors at an early age, often colon and brain tumors.[8]
Epigenetic silencing of mismatch repair genes[edit]
Sporadic cancers with a DNA repair deficiency only rarely have a mutation in a DNA repair gene, but they instead tend to have epigenetic alterations such as promoter methylation that inhibit DNA repair gene expression.[9] About 13% of colorectal cancers are deficient in DNA mismatch repair, commonly due to loss of MLH1 (9.8%), or sometimes MSH2, MSH6 or PMS2 (all ≤1.5%).[10] For most MLH1-deficient sporadic colorectal cancers, the deficiency was due to MLH1 promoter methylation.[10] Other cancer types have higher frequencies of MLH1 loss (see table below), which are again largely a result of methylation of the promoter of the MLH1 gene. A different epigenetic mechanism underlying MMR deficiencies might involve over-expression of a microRNA, for example miR-155 levels inversely correlate with expression of MLH1 or MSH2 in colorectal cancer.[11]
| Cancer type | Frequency of deficiency in cancer | Frequency of deficiency in adjacent field defect |
|---|---|---|
| Stomach | 32%[12][13] | 24%-28% |
| Stomach (foveolar type tumors) | 74%[14] | 71% |
| Stomach in high-incidence Kashmir Valley | 73%[15] | 20% |
| Esophageal | 73%[16] | 27% |
| Head and neck squamous cell carcinoma (HNSCC) | 31%-33%[17][18] | 20%-25% |
| Non-small cell lung cancer (NSCLC) | 69%[19] | 72% |
| Colorectal | 10%[10] |
MMR failures in field defects[edit]
A field defect (field cancerization) is an area of epithelium that has been preconditioned by epigenetic or genetic changes, predisposing it towards development of cancer. As pointed out by Rubin » …there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion.»[20][21] Similarly, Vogelstein et al.[22] point out that more than half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of apparently normal cells.
MLH1 deficiencies were common in the field defects (histologically normal tissues) surrounding tumors; see Table above. Epigenetically silenced or mutated MLH1 would likely not confer a selective advantage upon a stem cell, however, it would cause increased mutation rates, and one or more of the mutated genes may provide the cell with a selective advantage. The deficientMLH1 gene could then be carried along as a selectively near-neutral passenger (hitch-hiker) gene when the mutated stem cell generates an expanded clone. The continued presence of a clone with an epigenetically repressed MLH1 would continue to generate further mutations, some of which could produce a tumor.
MMR components in humans[edit]
In humans, seven DNA mismatch repair (MMR) proteins (MLH1, MLH3, MSH2, MSH3, MSH6, PMS1 and PMS2) work coordinately in sequential steps to initiate repair of DNA mismatches.[23] In addition, there are Exo1-dependent and Exo1-independent MMR subpathways.[24]
Other gene products involved in mismatch repair (subsequent to initiation by MMR genes) in humans include DNA polymerase delta, PCNA, RPA, HMGB1, RFC and DNA ligase I, plus histone and chromatin modifying factors.[25][26]
In certain circumstances, the MMR pathway may recruit an error-prone DNA polymerase eta (POLH). This happens in B-lymphocytes during somatic hypermutation, where POLH is used to introduce genetic variation into antibody genes.[27] However, this error-prone MMR pathway may be triggered in other types of human cells upon exposure to genotoxins [28] and indeed it is broadly active in various human cancers, causing mutations that bear a signature of POLH activity.[29]
MMR and mutation frequency[edit]
Recognizing and repairing mismatches and indels is important for cells because failure to do so results in microsatellite instability (MSI) and an elevated spontaneous mutation rate (mutator phenotype). In comparison to other cancer types, MMR-deficient (MSI) cancer has a very high frequency of mutations, close to melanoma and lung cancer,[30] cancer types caused by much exposure to UV radiation and mutagenic chemicals.
In addition to a very high mutation burden, MMR deficiencies result in an unusual distribution of somatic mutations across the human genome: this suggests that MMR preferentially protects the gene-rich, early-replicating euchromatic regions.[31] In contrast, the gene-poor, late-replicating heterochromatic genome regions exhibit high mutation rates in many human tumors.[32]
The histone modification H3K36me3, an epigenetic mark of active chromatin, has the ability to recruit the MSH2-MSH6 (hMutSα) complex.[33] Consistently, regions of the human genome with high levels of H3K36me3 accumulate less mutations due to MMR activity.[29]
Loss of multiple DNA repair pathways in tumors[edit]
Lack of MMR often occurs in coordination with loss of other DNA repair genes.[9] For example, MMR genes MLH1 and MLH3 as well as 11 other DNA repair genes (such as MGMT and many NER pathway genes) were significantly down-regulated in lower grade as well as in higher grade astrocytomas, in contrast to normal brain tissue.[34] Moreover, MLH1 and MGMT expression was closely correlated in 135 specimens of gastric cancer and loss of MLH1 and MGMT appeared to be synchronously accelerated during tumor progression.[35]
Deficient expression of multiple DNA repair genes is often found in cancers,[9] and may contribute to the thousands of mutations usually found in cancers (see Mutation frequencies in cancers).
Aging[edit]
A popular idea, that has failed to gain significant experimental support, is the idea that mutation, as distinct from DNA damage, is the primary cause of aging. Mice defective in the mutL homolog Pms2 have about a 100-fold elevated mutation frequency in all tissues, but do not appear to age more rapidly.[36] These mice display mostly normal development and life, except for early onset carcinogenesis and male infertility.
See also[edit]
- Base excision repair
- Nucleotide excision repair
References[edit]
- ^ Iyer RR, Pluciennik A, Burdett V, Modrich PL (February 2006). «DNA mismatch repair: functions and mechanisms». Chemical Reviews. 106 (2): 302–23. doi:10.1021/cr0404794. PMID 16464007.
- ^ Larrea AA, Lujan SA, Kunkel TA (May 2010). «SnapShot: DNA mismatch repair». Cell. 141 (4): 730–730.e1. doi:10.1016/j.cell.2010.05.002. PMID 20478261. S2CID 26969788.
- ^ Heller RC, Marians KJ (December 2006). «Replisome assembly and the direct restart of stalled replication forks». Nature Reviews. Molecular Cell Biology. 7 (12): 932–43. doi:10.1038/nrm2058. PMID 17139333. S2CID 27666329.
- ^ Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P (September 2010). «PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair». Proceedings of the National Academy of Sciences of the United States of America. 107 (37): 16066–71. doi:10.1073/pnas.1010662107. PMC 2941292. PMID 20713735.
- ^ Kadyrov FA, Dzantiev L, Constantin N, Modrich P (July 2006). «Endonucleolytic function of MutLalpha in human mismatch repair». Cell. 126 (2): 297–308. doi:10.1016/j.cell.2006.05.039. PMID 16873062. S2CID 15643051.
- ^ Flores-Rozas H, Clark D, Kolodner RD (November 2000). «Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex». Nature Genetics. 26 (3): 375–8. doi:10.1038/81708. PMID 11062484. S2CID 20861705.
- ^ Iyer RR, Pohlhaus TJ, Chen S, Hura GL, Dzantiev L, Beese LS, Modrich P (May 2008). «The MutSalpha-proliferating cell nuclear antigen interaction in human DNA mismatch repair». The Journal of Biological Chemistry. 283 (19): 13310–9. doi:10.1074/jbc.M800606200. PMC 2423938. PMID 18326858.
- ^ Online Mendelian Inheritance in Man (OMIM): 276300
- ^ a b c Bernstein C, Bernstein H (May 2015). «Epigenetic reduction of DNA repair in progression to gastrointestinal cancer». World Journal of Gastrointestinal Oncology. 7 (5): 30–46. doi:10.4251/wjgo.v7.i5.30. PMC 4434036. PMID 25987950.
- ^ a b c Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). «Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer». Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ^ Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. (April 2010). «Modulation of mismatch repair and genomic stability by miR-155». Proceedings of the National Academy of Sciences of the United States of America. 107 (15): 6982–7. Bibcode:2010PNAS..107.6982V. doi:10.1073/pnas.1002472107. PMC 2872463. PMID 20351277.
- ^ Kupčinskaitė-Noreikienė R, Skiecevičienė J, Jonaitis L, Ugenskienė R, Kupčinskas J, Markelis R, et al. (2013). «CpG island methylation of the MLH1, MGMT, DAPK, and CASP8 genes in cancerous and adjacent noncancerous stomach tissues». Medicina. 49 (8): 361–6. doi:10.3390/medicina49080056. PMID 24509146.
- ^ Waki T, Tamura G, Tsuchiya T, Sato K, Nishizuka S, Motoyama T (August 2002). «Promoter methylation status of E-cadherin, hMLH1, and p16 genes in nonneoplastic gastric epithelia». The American Journal of Pathology. 161 (2): 399–403. doi:10.1016/S0002-9440(10)64195-8. PMC 1850716. PMID 12163364.
- ^ Endoh Y, Tamura G, Ajioka Y, Watanabe H, Motoyama T (September 2000). «Frequent hypermethylation of the hMLH1 gene promoter in differentiated-type tumors of the stomach with the gastric foveolar phenotype». The American Journal of Pathology. 157 (3): 717–22. doi:10.1016/S0002-9440(10)64584-1. PMC 1949419. PMID 10980110.
- ^ Wani M, Afroze D, Makhdoomi M, Hamid I, Wani B, Bhat G, et al. (2012). «Promoter methylation status of DNA repair gene (hMLH1) in gastric carcinoma patients of the Kashmir valley» (PDF). Asian Pacific Journal of Cancer Prevention. 13 (8): 4177–81. doi:10.7314/apjcp.2012.13.8.4177. PMID 23098428.
- ^ Chang Z, Zhang W, Chang Z, Song M, Qin Y, Chang F, et al. (January 2015). «Expression characteristics of FHIT, p53, BRCA2 and MLH1 in families with a history of oesophageal cancer in a region with a high incidence of oesophageal cancer». Oncology Letters. 9 (1): 430–436. doi:10.3892/ol.2014.2682. PMC 4246613. PMID 25436004.
- ^ Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). «Head and neck squamous cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 gene». American Journal of Otolaryngology. 32 (6): 528–36. doi:10.1016/j.amjoto.2010.11.005. PMID 21353335.
- ^ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, et al. (October 2009). «Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma». Otolaryngology–Head and Neck Surgery. 141 (4): 484–90. doi:10.1016/j.otohns.2009.07.007. PMID 19786217. S2CID 8357370.
- ^ Safar AM, Spencer H, Su X, Coffey M, Cooney CA, Ratnasinghe LD, et al. (June 2005). «Methylation profiling of archived non-small cell lung cancer: a promising prognostic system». Clinical Cancer Research. 11 (12): 4400–5. doi:10.1158/1078-0432.CCR-04-2378. PMID 15958624.
- ^ Rubin H (March 2011). «Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture». BioEssays. 33 (3): 224–31. doi:10.1002/bies.201000067. PMID 21254148. S2CID 44981539.
- ^ Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, et al. (February 2000). «Genetic reconstruction of individual colorectal tumor histories». Proceedings of the National Academy of Sciences of the United States of America. 97 (3): 1236–41. Bibcode:2000PNAS…97.1236T. doi:10.1073/pnas.97.3.1236. PMC 15581. PMID 10655514.
- ^ Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). «Cancer genome landscapes». Science. 339 (6127): 1546–58. Bibcode:2013Sci…339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- ^ Pal T, Permuth-Wey J, Sellers TA (August 2008). «A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer». Cancer. 113 (4): 733–42. doi:10.1002/cncr.23601. PMC 2644411. PMID 18543306.
- ^ Goellner EM, Putnam CD, Kolodner RD (August 2015). «Exonuclease 1-dependent and independent mismatch repair». DNA Repair. 32: 24–32. doi:10.1016/j.dnarep.2015.04.010. PMC 4522362. PMID 25956862.
- ^ Li GM (January 2008). «Mechanisms and functions of DNA mismatch repair». Cell Research. 18 (1): 85–98. doi:10.1038/cr.2007.115. PMID 18157157.
- ^ Li GM (July 2014). «New insights and challenges in mismatch repair: getting over the chromatin hurdle». DNA Repair. 19: 48–54. doi:10.1016/j.dnarep.2014.03.027. PMC 4127414. PMID 24767944.
- ^ Chahwan R, Edelmann W, Scharff MD, Roa S (August 2012). «AIDing antibody diversity by error-prone mismatch repair». Seminars in Immunology. 24 (4): 293–300. doi:10.1016/j.smim.2012.05.005. PMC 3422444. PMID 22703640.
- ^ Hsieh P (September 2012). «DNA mismatch repair: Dr. Jekyll and Mr. Hyde?». Molecular Cell. 47 (5): 665–6. doi:10.1016/j.molcel.2012.08.020. PMC 3457060. PMID 22980456.
- ^ a b Supek F, Lehner B (July 2017). «Clustered Mutation Signatures Reveal that Error-Prone DNA Repair Targets Mutations to Active Genes». Cell. 170 (3): 534–547.e23. doi:10.1016/j.cell.2017.07.003. hdl:10230/35343. PMID 28753428.
- ^ Tuna M, Amos CI (November 2013). «Genomic sequencing in cancer». Cancer Letters. 340 (2): 161–70. doi:10.1016/j.canlet.2012.11.004. PMC 3622788. PMID 23178448.
- ^ Supek F, Lehner B (May 2015). «Differential DNA mismatch repair underlies mutation rate variation across the human genome». Nature. 521 (7550): 81–4. Bibcode:2015Natur.521…81S. doi:10.1038/nature14173. PMC 4425546. PMID 25707793.
- ^ Schuster-Böckler B, Lehner B (August 2012). «Chromatin organization is a major influence on regional mutation rates in human cancer cells». Nature. 488 (7412): 504–7. Bibcode:2012Natur.488..504S. doi:10.1038/nature11273. PMID 22820252. S2CID 205229634.
- ^ Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (April 2013). «The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα». Cell. 153 (3): 590–600. doi:10.1016/j.cell.2013.03.025. PMC 3641580. PMID 23622243.
- ^ Jiang Z, Hu J, Li X, Jiang Y, Zhou W, Lu D (December 2006). «Expression analyses of 27 DNA repair genes in astrocytoma by TaqMan low-density array». Neuroscience Letters. 409 (2): 112–7. doi:10.1016/j.neulet.2006.09.038. PMID 17034947. S2CID 54278905.
- ^ Kitajima Y, Miyazaki K, Matsukura S, Tanaka M, Sekiguchi M (2003). «Loss of expression of DNA repair enzymes MGMT, hMLH1, and hMSH2 during tumor progression in gastric cancer». Gastric Cancer. 6 (2): 86–95. doi:10.1007/s10120-003-0213-z. PMID 12861399.
- ^ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). «Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2». Proceedings of the National Academy of Sciences of the United States of America. 94 (7): 3122–7. Bibcode:1997PNAS…94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
Further reading[edit]
- Hsieh P, Yamane K (2008). «DNA mismatch repair: molecular mechanism, cancer, and ageing». Mechanisms of Ageing and Development. 129 (7–8): 391–407. doi:10.1016/j.mad.2008.02.012. PMC 2574955. PMID 18406444.
- Iyer RR, Pluciennik A, Burdett V, Modrich PL (February 2006). «DNA mismatch repair: functions and mechanisms». Chemical Reviews. 106 (2): 302–23. doi:10.1021/cr0404794. PMID 16464007.
- Joseph N, Duppatla V, Rao DN (2006). Prokaryotic DNA mismatch repair. Progress in Nucleic Acid Research and Molecular Biology. Vol. 81. pp. 1–49. doi:10.1016/S0079-6603(06)81001-9. ISBN 9780125400817. PMID 16891168.
- Yang W (August 2000). «Structure and function of mismatch repair proteins». Mutation Research. 460 (3–4): 245–56. doi:10.1016/s0921-8777(00)00030-6. PMID 10946232.
- Griffiths JF, Gilbert WM, Lewontin RC, Wessler SR, Suzuki DT, Miller JH (2004). An introduction to genetic analysis (8th ed.). New York, NY: Freeman. ISBN 978-0-7167-4939-4.
- Kunkel TA, Erie DA (2005). «DNA mismatch repair». Annual Review of Biochemistry. 74: 681–710. doi:10.1146/annurev.biochem.74.082803.133243. PMID 15952900.
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger (2005). DNA repair and mutagenesis (2nd ed.). Washington, D.C.: ASM Press. ISBN 978-1-55581-319-2.
External links[edit]
- DNA Repair Archived 2018-02-12 at the Wayback Machine
- DNA+Mismatch+Repair at the U.S. National Library of Medicine Medical Subject Headings (MeSH)