
Индикаторные погрешности титрования
При титровании возможны случайные и
систематические погрешности. Случайные
погрешности связаны с измерением объема
и массы навески, но и значительную часть
погрешности титрования составляют
систематические погрешности, в частности,
индикаторная.
Случайные погрешности обрабатываются
по законам математической статистики.
Индикаторные погрешности связаны с
тем, что pT индикатора не
совпадает со значением pH
в ТЭ. Конечная точка титрования с данным
индикатором не совпадает с ТЭ.
При недотитровывании:
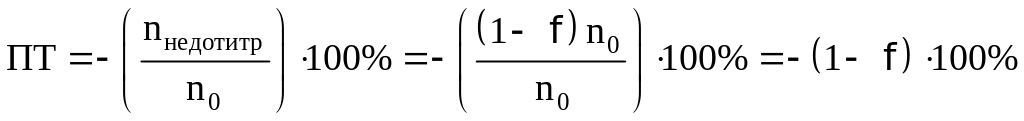
При перетитровывании:
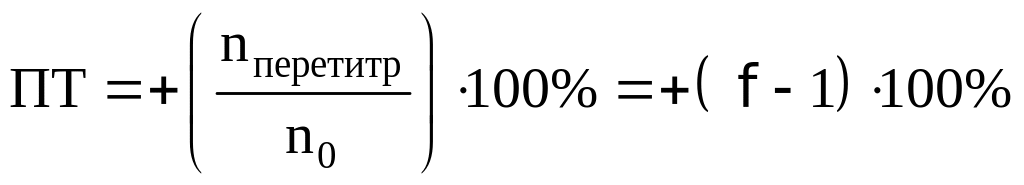
Возьмем индикаторы хризоидин (pT=5,50)
и хлорфеноловый красный (pT
= 5,80). В данном случае в КТТ pH
будет больше, чем pH в ТЭ
(pH = 5,28), а, следовательно,
в растворе будет неотитрованное
основание. Эта погрешность, обусловленная
содержанием неоттитрованного основания,
называется щелочной, будет определяться
уравнением
![]()
.
На данном этапе титрования pH
будет определяться по формуле
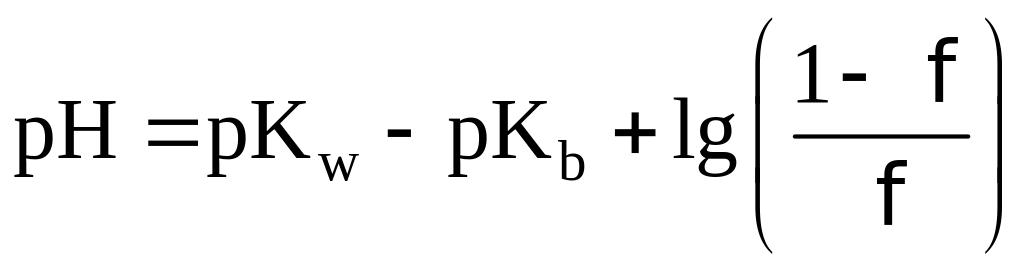
.
В КТТ pH раствора равен
pT. Следовательно, можно
найти и f
в КТТ:
![]()
.
Отсюда,

.
Посчитаем ПТ для данных индикаторов:
хризоидин:
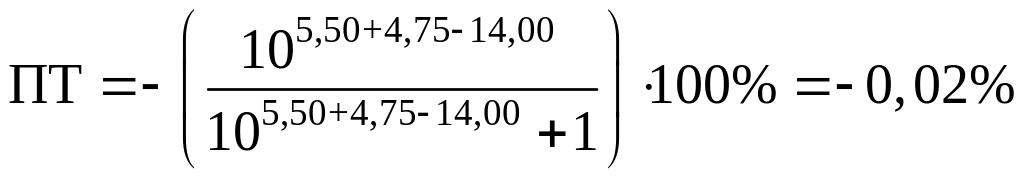
хлорфеноловый красный:
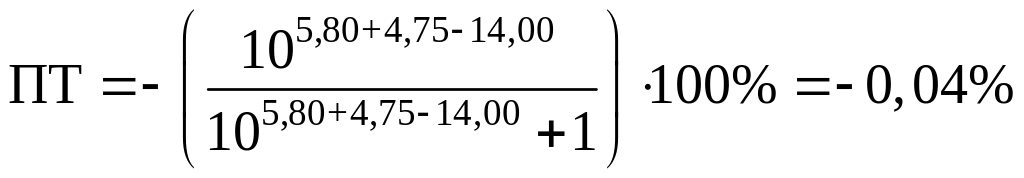
Оба эти индикатора подойдут для нашего
титрования. Рассмотрим ПТ для индикатора
розоловая кислота (pT=7,1).
По данной формуле получается ПТ=–0,70%,
что превышает обычно задаваемое значение
погрешности (±0,2%).
Рассмотрим же теперь случай, когда мы
используем индикаторы с pT
меньшим, чем pH в ТЭ. В КТТ
раствор будет перетитрован, и pH
будет определяться концентрацией
сильной кислоты (водородная погрешность),
и в нашей задаче определяться уравнением
.
Погрешность будет определяться по
формуле
![]()
Возьмем для примера индикаторы лакмоид
(pT=5,20), ализариновый красный
C (pT=4,45) и
бромфеноловый синий (pT=3,80).
pH раствора в КТТ равен
pT:
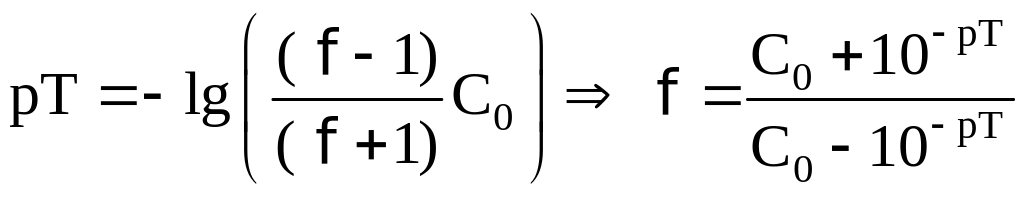
.
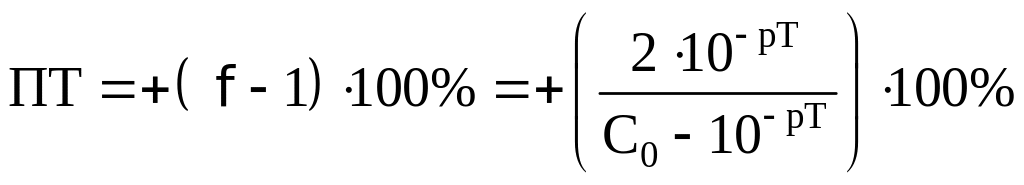
.
Рассчитаем ПТ для наших индикаторов:
Лакмоид:

Ализариновый красный C:
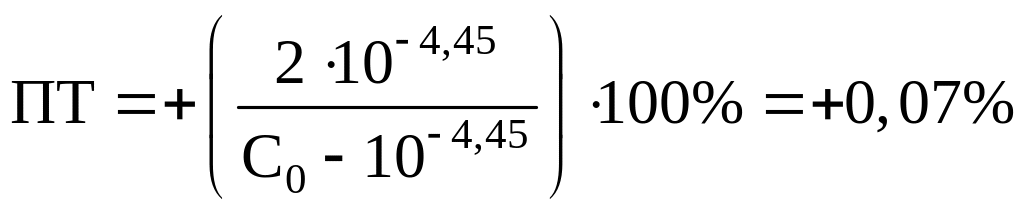
Бромфеноловый синий:

Вполне очевидно, что из двух предложенных
индикаторов наиболее подходящим является
ализариновый красный C.
Учитывая все расчеты, приходим к выводу,
что самыми подходящими для нашего опыта
индикаторами являются хризоидин (с
интервалом перехода 4,0 – 7,0, оранжевая
– желтая) и лакмоид (4,0 – 6,4, красная –
синяя).
Выводы
По кривой титрования аммиака можно
сделать ряд выводов.
В ходе титрования заметно плавное
уменьшение pH и заметен
скачок в области точки эквивалентности.
Скачок титрования полностью находится
в кислой области.
Точка эквивалентности расположена при
pH 5,28 и, очевидно, не
совпадает с точкой нейтральности. Скачок
титрования 0,1 М аммиака в пределах ±0,1%
от точки эквивалентности находится в
пределах pH от 6,25 до 4,30 и
составляет примерно 2 единицы pH,
что намного меньше скачка сильной щелочи
(6 единиц pH). С уменьшением
концентрации и увеличением температуры
скачок уменьшается.
В нашем случае одними из самых подходящих
являются лакмоид и хризоидин.
Окислительно-восстановительное титрование
Метод основан на реакциях
окисления-восстановления. Их называют
по применяемому тированному раствору
реагента, например: перманганатометрия,
йодометрия, бихроматометрия. В этих
методах в качестве титрантов применяют,
соответственно, KMnO4,
I2, K2Cr2O7.
В основе метода лежит изменение
окислительно-восстановительного
потенциала, обусловленного протеканием
окислительно-восстановительной реакции
между титрантом и определяемым веществом.
![]()
В процессе титрования происходит
изменение концентраций окисленной и
восстановленной форм, а, следовательно,
изменяется окислительно-восстановительный
потенциал титруемого раствора, включающей
две редоксопары.
В соответствии с уравнением Нернста
окислительно-восстановительный потенциал
для любой редоксопары:
![]()
Для каждого отдельного метода
окислительно-восстановительного
титрования используются свои стандартные
растворы.
Рассмотрим наш случай – перманганатометрия.
Рабочим раствором этого метода является
раствор перманганата калия KMnO4,
он неустойчив из-за реакции с водой,
катализируемый диоксидом марганца и
на свету:
![]()
Поэтому растворы перманганата калия
следует готовить, используя чистую воду
(органические примеси в воде могут
реагировать с
![]()
и давать MnO2, ускоряющий
разложение реагента), отфильтровать от
диоксида марганца и хранить в темных
склянках; раствор следует выдержать
несколько недель для окончания протекания
всех процессов. Очевидно, что раствор
следует стандартизировать, для чего
используют оксалат натрия и другие
восстановители. Реакция
![]()
катализируется ионами Mn2+.
Первые капли перманганата даже в горячем
растворе обесцвечиваются очень медленно.
В ходе титрования концентрация ионов
Mn2+ возрастает и
скорость реакции увеличивается: реакция
автокаталитическая.
Титр перманганата калия можно установить
также по оксиду мышьяка(III)
или металлическому железу.
В перманганатометрии применяют также
растворы восстановителей – слои Fe(II),
щавелевую кислоту и некоторые другие
– для определения окислителей методом
обратного титрования. Соединения Fe(II)
на воздухе медленно окисляются, особенно
в нейтральном растворе. Подкисление
замедляет процесс окисления, однако
обычно рекомендуется перед применением
раствора Fe(II)
в анализе проверить его титр. Оксалаты
и щавелевая кислота в растворе медленно
разлагаются. Этот процесс ускоряется
на свету, поэтому растворы оксалатов
рекомендуется хранить в темных склянках.
Подкисленные растворы оксалатов более
устойчивы, чем нейтральные или щелочные.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Чтобы окраска окислительно-восстановительного индикатора изменялась при титровании резко и индикаторная ошибка титрования была незначительной, интервал перехода индикатора должен находиться в пределах скачка потенциалов на кривой титрования. [c.369]
И. М. Кольтгоф, В. А. Стенгер. Объемный анализ. Госхимиздат, 1950, (т. I. 376 стр.) и 1952, (т. И, 444 стр.). В т. I рассматриваются теоретические основы объемного анализа. Изложена теория методов нейтрализации и соединения ионов, приведены кривые титрования для различных случаев метода нейтрализации. Отдельные главы содержат материал ио теории методов окисления-восстановления, теории индикаторов, по ошибкам титрования. Рассмотрены явления адсорбции и соосаждения, катализа и индукции, применение объемных методов в органическом анализе описаны теоретические положения, касающиеся применения физико-химических методов для определения точки эквивалентности. В т. 11 книги изложено практическое применение методов нейтрализации, осаждения и комплексообразования. В томе 111 (840 стр., 1961 г.) описано применение окислительно-восстановительных методов объемного анализа. [c.486]
Инцикаторы в окислительно-восстановительном титровании. Индикаторные ошибки титрования [c.132]
Ошибка титрования близка нулю, если потенциал перехода окраски индикатора равен потенциалу в точке эквивалентности. Практически допустимой считают относительную ошибку в10 или 0,1 7о, благодаря чему возможен широкий выбор окислительно-восстановительных индикаторов. При титровании восстановителя 2 окислителем 1 предельные значения потенциа-.лов (в вольтах) получают из следующих уравнений [c.170]
Кривые титрования хлоридов нитратом серебра в расплавленных солях получаются такими же, как кривые титрования в водных растворах для экзотермических процессов. Относительная ошибка титрования для указанных выше концентраций приближалась к 4%. Применение метода ограничено в связи с трудностью достаточно точного измерения повышения температуры, происходящего в результате реакции. Необходимо, чтобы изменение температуры окружающей среды было на несколько порядков меньше, чем изменение температуры, происходящее в результате рассматриваемой реакции. Теоретически в среде расплавленных солей возможно проводить любые реакции осаждения, комплексообразования или окислительно-восстановительные реакции, однако практически сложность прибора, который требуется для этого, ограничивает применимость расплавленных солей в качестве растворителей. [c.110]
На практике расчет потенциала при помощи такого уравнения, как правило, связан с ошибкой, хотя во многих случаях эта ошибка невелика и полученное значение потенциала является полезным для предсказания возможности проведения того или иного титрования. Во всех окислительно-восстановительных системах, включающих ионы водорода, потенциал обычно зависит от концентрации этих ионов, однако степень такой зависимости может значительно отличаться от величины, полученной по уравнению. Такая же картина иногда наблюдается и для зависимости потенциала от концентрации окисленной и (или) восстановленной формы вещества. [c.353]
Разница в объемах постоянна для всех выбранных на кривой точек, до которых проводилось титрование, и является, вероятно, приборной ошибкой. Результаты титрования до заданного потенциала показывают, что при помощи автотитратора можно приготавливать растворы с заранее заданными величинами окислительно-восстановительного потенциала или pH, что позволяет автоматизировать процесс подготовки проб к дальнейшему анализу при серийных определениях. [c.136]
Для окислительно-восстановительного титрования перманганата применяют также другие восстановители, в том числе соединения Си, Sn , W», U , V [17], однако преимущества применения этих восстановителей не очевидны. Сравнительно простой окислительно-восстановительный метод основан на взаимодействии перманганата с кислым раствором KI и последующем титровании выделившегося иода стандартным раствором тиосульфата. Избирательность определения часто достигается применением маскировки. Например, в указанном выше методе перманганат (или хромат) можно определять в присутствии железа (III), которое маскируют гексаметафосфатом [18]. При определении 13—65 мг марганца (в виде MnO ) в присутствии 10—340 мг Fe + ошибка составляет 0,3%. [c.159]
В Присутствии восстановителей-абсорбентов в ячейке для поглощения. Осуществить такое определение с хлор- или бромсеребряными электродами не представляется возможным, так как они дают ошибку, связанную с возникновением на них окислительно-восстановительного потенциала. Сжигание проводится обычным способом, потенциометрическое титрование идет в 50%-ном диоксане. Титруют 0,02 М раствором нитрата серебра. Вблизи конечной точки титрант вводят равными порциями по 0,02 мл. Конечную точку определяют как максимальное значение Д /Д . [c.62]
Амперометрическое титрование применимо для многих окислительно-восстановительных реакций, реакций комплексообразования и осаждения. По сути этот метод обладает более высокой точностью, чем соответствующий прямой полярографический метод, так как каждое определение включает ряд отдельных измерений, для которых исключаются случайные ошибки. [c.359]
Преимущество флуоресцентных окислительно-восстановительных индикаторов по сравнению с цветными состоит в том, что с их применением можно значительно уменьшить ошибки титрования, связанные с расходом титранта на окисление индикатора. Кроме того, возможно титрование в окрашенных и мутных средах . [c.120]
Окислительно-восстановительные реакции в большинстве случаев протекают в сильно кислой среде небольшие изменения электропроводности за счет реакции окисления-восстановления на фоне высокой проводимости среды обусловливают нерезкие перегибы кривой титрования в конечной точке. Это приводит к значительным ошибкам в определении конечной точки. В работе [69] систематизированы реакции этого типа в наглядной таблице для целей кондуктометрического титрования. Эта таблица может быть использована при разработке методов ВЧ-титрования окислителей и восстановителей. [c.156]
Последние соотнощения показывают, что в состоянии равновесия концентрация Мп2+ будет превыщать концентрацию МпО в 1025 раз . р g процесс восстановления ионов МпО в Мп2+ пройдет с исчерпывающей для практической цели полнотой. Поэтому можно считать, что ощибка титрования, обусловленная образованием равновесной системы, будет равна нулю (такое количественное течение процесса вообще характерно для большинства окислительно-восстановительных реакций). Ввиду этого ошибки, допускаемые при перманганатометрических определениях, должны быть отнесены лишь за счет капельной ошибки, неточностей при отсчетах и взвешиваниях, а также необходимости введения некоторого избытка перманганата, позволяющего установить конец реакции по появлению окраски раствора. [c.85]
Практикум содержит 47 лабораторных заданий. Многие задания оригинальны по постановке и рассчитаны на получение количественных результатов. Некоторые из задач могут быть использованы как факультативные. Новые работы посвящены методам очистки веществ ионитами и зонной плавкой, определению термодинамических характеристик процесса растворения бензойной кислоты и процесса восстановления ионов меди цинком, определению координационного числа методами криоскопии и титрования, кинетике окислительно-восстановительных реакций и изучению поверхностно-активных веществ. Заново написано задание по определению тепловых эффектов. В новом издании практикума лабораторным заданиям предпослано введение, где рассказывается о научном эксперименте и его роли в познании, и глава о работе с экспериментальными данными, в которой идет речь о записи результатов, вычислениях, ошибках эксперимента, выражении результатов в виде графиков и формул и о написании отчета. Остальные задания практикума подверглись значительной переработке. В приложении появился ряд новых таблиц. [c.3]
В книге на современном уровне кратко изложены теоретические основы гравиметрии и титриметрии — образование и свойства осадков, типы химических равновесий в гомогенных и гетерогенных растворах описаны кривые титрования проанализированы ошибки в кислотно-основном, осацительном, комплексимет-рическом и окислительно-восстановительном титровании. Подробно рассмотрены аппаратура и техника проведения всех операций в количественном химическом анализе. Все расчеты проведены с учетом новых данных о величинах констант, стандартных потенциалов и т.п. [c.2]
Окислительно-восстановительный потенциал аскорбиновой кислоты при pH 7 составляет — -0,19 в. Аскорбинометрическое определение железа является одним из лучших методов, так как не мешают нитраты и фосфаты присутствие фторидов вызывает незначительную ошибку. Недостатком является малая устойчивость титрованного раствора аскорбиновой кислоты при хранении. [c.193]
Кинетическое окислительно-восстановительное титрование Sb(lII). Реакционную смесь титруют раствором окислителя (КВгОз, Ja, e(S04)2, K rjO,, KJO3) в строго определенных условиях (pH, температура, объем) при постоянной скорости подачи титранта с потенциометрическим, фотометрическим или визуальным (индикаторы ксиленоловый оранжевый, ферроин) установлением конечной точки. По продолжительности титрования, которое прямо пропорционально содержанию Sb, находят ее содержание. Метод позволяет определять Sb в растворах с ее концентрацией 8-10 —1,2-10 с ошибкой 2—5% [953, 1326]. [c.98]
Эрдеи и Ради [938] титровали Au(III) аскорбиновой кислотой при pH 1—3 и 50—60° С. При температуре > 80° С получаются заниженные результаты. Не мешают Hg(II), u, Fe(III) (в присутствии НэР04), 150-кратные количества NOJ, 1 г-ион л С мешают ионы со стандартным окислительно-восстановительным потенциалом > — — 1,39 в [Pt(IV), Вг , S N и N ]. Ошибка определения золота в 0,001—0,01 N растворах 1%. При титровании в среде ледяной уксусной кислоты [937] вид кривой титрования похож на кривую титрования в водных растворах, если перед титрованием в безводной уксусной кислоте ввести безводный Ha OONa. В точке эквивалентности наблюдается отчетливый скачок потенциала. Аналогично золоту ведут себя другие окислители. [c.130]
ВОЗМОЖНОЙ благодаря снижению окислительно-восстановительного потенциала пары Fe +/Fe + за счет образования устойчивых пирофосфатных комплексов Ее(П1) при pH 9, а в начале титрования — при pH 12 [915]. Титрование ведут 0,01 JV раствором ЕеЗОд в инертной атмосфере с обязательным добавлением в анализируемый раствор бромид-иона, препятствующего диспропорцио-нированию гипобромит-иона. 1 —10 мг ВгО определяют этим методом с ошибкой -< 0,5%. Примеси BrOJ (до 16%), 10-кратное количество ионов СГ или J , 10-кратный избыток сульфат-иона и 150-кратный нитрат-иона не мешают анализу. [c.129]
При прямом титровании фосфатов раствором соли свинца [1172] при рн 2—3 в качестве индикатора применяют хлороформный раствор дитизона. Титруют до перехода зеленой окраски в фиолетовую. Метод применяют для определения фосфора в фосфатных удобрениях [1174]. В качестве индикатора применяют также эриох-ром черный Т (растворяют 0,2 г эриохрома черного Т в 5 мл С2Н5ОН и 15 мл триэтаНоламина) [950]. Титруют до появления красной окраски. Метод применяют для определения микроколичеств фосфора в органических веществах. Для определения микроколичеств фосфора применяют также титрование нитратом свинца в присутствии 2-азо-4-резорцина [1018]. Титруют до появления красного окрашивания. При содержании фосфора 20— 400 мкг средняя абсолютная ошибка определения составляет 2—3 мкг Р. При косвенном определении фосфатов с помощью нитрата свинца применяют окислительно-восстановительные индикаторы [732, 733]. Метод основан на осаждении РО/ в виде РЬз(Р04)2 нитратом свинца, избыток которого оттитровывают К4[Ге(СК)б1 в присутствии вариаминового синего и Кз[Ге(СК)б] в качестве индикатора. Титруют до перехода фиолетовой окраски в бледно-желтую. [c.37]
Предложен интересный метод окислительно-восстановительного титрования, основанный на применении гидрохинона [20]. Метод позволяет определять 10—130 мг перманганата в присутствии других окислителей, например, бихромата, гексацианоферрата (III) и хлорамина Т. При определении 13—130 мг КМПО4 в присутствии 20—2000 мг других окислителей относительная ошибка оп-)еделения не превышает 1,8%. Определению мешает ванадий (V). Метод длителен сначала перманганат восстанавливают до диоксида марганца с помощью формиата натрия в щелочном растворе. Осадок гидратированного диоксида марганца фильтруют, промывают, растворяют в ЫагНгРгОу и образующийся пирофосфат марганца (III) титруют стандартным раствором гидрохинона. [c.159]
Хотя растворы бихромата окрашены в оранжевый цвет, интенсивность окраски недостаточна для определения конечной точки. Отличным индикатором при титровании бихроматом является дифениламиносульфокислота (гл. 15) с переходом окраски от зеленой (ионы хрома(1П)) до фиолетовой (окисленная форма индикатора). Холостой опыт в присутствии индикатора идет плохо, поскольку в отсутствие других окислительно-восстановительных систем бихромат очень медленно окисляет индикатор. Но ошибка, возникающая за счет того, что холостой опыт не проведен, обычно пренебрежимо мала. Реакция окисления дифениламиносульфокис-лоты обратима, поэтому можно проводить обратное титрование очень малых количеств бихромата железом (II). В присутствии больших количеств окислителя при низкой кислотности (рН>2) раствора индикатор необратимо окисляется до соединений желтого или красного цвета. [c.386]
Метод определения конечной точки. Как уже отмечалось, перманганат-ион имеет настолько интенсивный цвет, что сам по себе может служить индикатором. Опыты показали, что при титровании мышьяковистой кислоты перманганатом концентрация иона МпОГ. равная б-10 моль/л, легко обнаруживается вблизи точки эквивалентности. В расчетах, проведенных для одного из опытов титрования, мы показали, что при введении 0,1% избытка титранта концентрация МпОГ составляет 8-10 моль/л. Таким образом, ясно, что при использовании цвета избыточного Мп04 для установления точки эквивалентности вводится ошибка менее 0,1 %. Однако можно подобрать окислительно-восстановительный индикатор, при применении которого конечная точка еще точнее соответствовала бы точке эквивалентности. В рассматриваемом случае важнее даже то, что при использовании индикатора конечную точку можно обнаружить еще в тот момент, когда концентрация МпОГ слишком низка, чтобы окислить хлорид-ионы, присутствующие в растворе. Один из таких индикаторов — о-фенаитролиповый комплекс желе-за П), с которым могут происходить следующие превращения [c.235]
Работа установки проверялась не только титрованием в кислотно основных и окислительно-восстановительных сис темах, но также пу тем определения миллиграммовых количеств галогенид ионов их осаждением ионами серебра, г енерированнымн электрохимически. Определения проводят в бюксе на 75 мл, анодом служит серебро вы сокой чистоты или платина, покрытая серебром, катодом — платиновая спираль, снабженная чехлом. В качестве индикаторного электрода используют серебряную или платиновую проволоку, потенциал ко торой измеряется относительно каломельного электрода сравнения, причем для устранения загрязнения хлоридом последний под.-оединя-н.т с помощью агарового солевого мостика- с 0,1 М раствором а. 0,. При определении хлорида, бромида и иодида в количествах 0,2 — 10 средняя ошибка составляет 0,005 мг. [c.86]
1.
Окислительно-восстановительное
титрование
1
2.
Mэкв(X) = fэкв · М(X)
f = 1/z
z – число электронов, принимающих участие в
окислительно-восстановительной реакции
2
3.
10FeSO4 + 2KMnO4 + 8H2SO4 2MnSO4 +
5Fe2(SO4)3 + K2SO4 + 8H2O
MnO4 + 8H+ + 5ē Mn2+ + 4H2O
2Fe2+ 2ē Fe23+
3
4.
10FeSO4 + 2KMnO4 + 8H2SO4 2MnSO4 +
5Fe2(SO4)3 + K2SO4 + 8H2O
MnO4 + 8H+ + 5ē Mn2+ + 4H2O
2Fe2+ 2ē Fe23+
fэкв(KMnO4) = 1/5,
M(1/5KMnO4) = M(KMnO4) /5;
fэкв(FeSO4) = 1
M(FeSO4) = M(FeSO4)/1
4
5.
I2 + 2Na2S2O3 2NaI + Na2S4O6
I2 + 2ē 2I
2S2O32 2ē S4O62
5
6.
I2 + 2Na2S2O3 2NaI + Na2S4O6
I2 + 2ē 2I
2S2O32 2ē S4O62
fэкв(I2) = 1/2
M(1/2 I2) = M(I2)/2;
fэкв(Na2S2O3) = 1
M(Na2S2O3) = M(Na2S2O3)/1
6
7.
6KI + K2Cr2O7 + 7H2SO4 3I2 + Cr2(SO4)3 +
K2SO4 + 7H2O
Cr2O72 + 14H+ + 6ē Cr23+ + 7H2O
2I 2ē I2
7
8.
6KI + K2Cr2O7 + 7H2SO4 3I2 + Cr2(SO4)3 +
K2SO4 + 7H2O
Cr2O72 + 14H+ + 6ē Cr23+ + 7H2O
2I 2ē I2
fэкв(K2Cr2O7) = 1/6
Mэкв(K2Cr2O7) = M(K2Cr2O7)/6
fэкв(KI) = 1
M(KI) = M(KI)/1
8
9.
H2SO4
CH4
CH3—CH2OH
CH3OH
H
HCOH
H
H
CH3—COOH
H
H
H
9
10.
–4
–2
CH4
CH3OH
–3
–1
CH3—CH2OH
+1
HCOH
–3
-1
+3
CH3—COOH
10
11.
O O
OH OH
H
+1
H +2
+1
O
O
+2
CHOH
O
CHOH
CH2OH
CH2OH
C21+ – 2е
O
C22+
fэкв(Аск.к.) = 1/2
Mэкв(Аск.к.) = M(Аск.к.)/2
11
12.
Перманганатометрия
Титрант – 0,02 М или 0,1 М (1/5 KMnO4) или
0,1 н. раствор KMnO4
По точной навеске приготовить нельзя, т.к.
сильный окислитель.
Готовят раствор приблизительно нужной концентрации, выдерживают 7-10 дней или
кипятят 10 минут для окисления восстановителей, содержащихся в воде
Фильтруют через стеклянный фильтр.
12
13.
Стандартизацию проводят по щавелевой кислоте H2C2O4 2H2O, оксалату натрия Na2C2O4,
оксиду мышьяка (III) As2O3, металлическому
железу.
Стандартизацию проводят только в сернокислой
среде.
16HCl + 2KMnO4 5Cl2 + 2MnCl2 + 2KCl +8H2O
Индикатор – сам титрант.
13
14.
Стандартизация 0,1 н. по щавелевой кислоте
60-70 0С
5H2C2O4 + 2KMnO4 + 3H2SO4 2MnSO4 +
10CO2 + K2SO4 + 8H2O
C2O42– – 2е 2CO2
14
15.
Этикетка:
0,02 М или 0,1 М (1/5KMnO4) или 0,1 н.
К = 0,9972
Хранят в темном месте, в склянках темного
стекла
свет
4KMnO4 + 2H2O 4MnO2 + 4 KOH + 3H2O
По ГФ XIII стандартизация по тиосульфату
натрия
15
16.
Применение:
прямая перманганатометрия
Железа (II) сульфат:
10FeSO4 + 2KMnO4 + 8H2SO4 5Fe2(SO4)3 +
K2SO4 + 2MnSO4 + 8H2O
2Fe2+ – 2е Fe23+
Сэкв(KMnO4)·Mэкв(FeSO4)
Т(KMnO4/FeSO4) = ————————————
1000
16
17.
m(FeSO4) = V(KMnO4)·K·T(KMnO4/FeSO4)
V(KMnO4)·K·T(KMnO4/FeSO4)·100
(FeSO4) = ——————————————, %
а(FeSO4)
17
18.
Перекись водорода:
5H2O2 + 2KMnO4 + 3H2SO4 5O2 + K2SO4 +
2MnSO4 + 8H2O
O22– – 2е O2
Сэкв(KMnO4)·Mэкв(H2O2)
Т(KMnO4/H2O2) = ————————————
1000
18
19.
Аскорбиновая кислота:
O O
OH OH
H
H
5
+ 2KMnO4 + 3H2SO4
O
5
CHOH
O
CHOH
CH2OH
CH2OH
O
C21+ – 2е
+ 2MnSO4 + K2SO4 + 8H2O
O
C22+
Сэкв(KMnO4)·Mэкв(Аск.к.)
Т(KMnO4/Аск.к.) = ————————————
1000
19
20.
обратная перманганатометрия
Натрия нитрит:
5NaNO2 + 2KMnO4(изб.) + 3H2SO4 5NaNO3 +
2MnSO4 + K2SO4 + 3H2O
2KMnO4(ост.) + 10KI + 8H2SO4 2MnSO4 + 5I2
+ 6K2SO4 + 8H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
20
21.
(VKMnO4 K VNa2S2O3 K) Т(Na2S2O3/NaNO2) 100
(NaNO2)= (%)
а (NaNO2)
Сэкв(Na2S2O3) Мэкв(NaNO2)
Т(Na2S2O3/NaNO2) =
1000
NO2– + H2O – 2е
N3+ – 2е
NO3– + 2H+
N5+
21
22.
5NaNO2 + 2KMnO4(изб.) + 3H2SO4 5NaNO3 +
2MnSO4 + K2SO4 + 3H2O
NO2– + H2O – 2е NO3– + 2H+
| 5
MnO4– + 8H+ + 5е Mn2+ + 4H2O | 2
5NO2– + 2MnO4– + 16H+ + 5H2O
5NO3– + 10H+ + 2Mn2+ + 8H2O
или
5NO2– + 2MnO4– + 6H+ 5NO3– + 2Mn2+ + 3H2O
22
23.
Кривые окислительно-восстановительного
титрования
Выражают зависимость величины потенциала
от концентрации титранта
23
24.
Задача. Рассчитать и построить кривую титрования соли Fe (II) раствором перманганата
калия, если
Eo (Fe3+/Fe2+) = + 0,77 B;
Eo (MnO4–/Mn2+) = + 1,51 B
Какой из индикаторов можно использовать в
данном случае?
24
25.
2,2 – Дипиридил (комплекс с рутением)
Ео = 1,33 В
Дифениламин-2,2 –дикарбоновая кислота
Ео = 1,26 В
Комплекс 1,10-фенантролина с Fe (II)
Ео = 1,14 В
Фенилантраниловая кислота Ео = 1,08 В
Дифениламин
Ео = 0,76 В
Метиленовый голубой
Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
25
26.
5Fe2+ + MnO4– + 8H+ 5Fe3+ + Mn2+ + 4H2O
В начале титрования и до т.э. в растворе будут:
Fe2+, Fe3+ ,Mn2+
Потенциал системы будет определяться парой
восстановителя Fe3+/Fe2+
Величина потенциала может быть вычислена по
уравнения Нернста:
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
26
27.
В точке эквивалентности:
E = (mEoв-ля + nEoок-ля )/(m + n)
27
28.
5Fe2+ + MnO4– + 8H+ 5Fe3+ + Mn2+ + 4H2O
за т. э. в растворе будут:
Fe3+, MnO4– и Mn2+
Потенциал будет определяться парой
окислителя MnO4– /Mn2+
Величина потенциала вычисляется по
уравнению Нернста:
E = Eo + 0,059/5 lg[MnO4–][H+]8/[Mn2+]
28
29.
Пусть концентрация ионов водорода равна 1 М
Доб-но
От-но
KMnO4–
в%
[Fe3+]
———
[Fe2+]
9
9
9/91 10-1
-1
0,77–0,059=0,71В
50
50
50/50=1
0
0,77 В
99
99
99/1 102
2
0,77+0,059 2=0,89В
3
0,77+0,059 3=0,95В
99,9
100
99,9 99,9/0,1 103
[Fe3+]
lg ———
[Fe2+]
E=Eo +0,059/1
lg [Fe3+]/[Fe2+]
E = (mEoв-ля+nEoок-ля )/(m+n)=(1 0,77+5 1,51)/(1+5)
=1,39 29B
30.
Доб-но
Изб-к
KMnO4–
в%
100,1
0,1
[MnO4–][H+]8
————— lgдроби E=Eo +0,059/5lgдр
[Mn2+]
0,1/100 10-3 -3
1,51+(0,059/5) (-3) =
1,475 B
101
1
1/100=10-2
-2
1,51+(0,059/5) (-2)
= 1,486 B
110
10
10/100=10-1
-1
1,51+(0,059/5) (-1)
= 1,498 B
30
31.
Скачок от 0,95 В до 1,475 В
2,2 – Дипиридил (комплекс с рутением)
Ео = 1,33 В
Дифениламин-2,2 –дикарбоновая кислота
Ео = 1,26 В
Комплекс 1,10-фенантролина с Fe (II)
Ео = 1,14 В
Фенилантраниловая кислота Ео = 1,08 В
Дифениламин Ео = 0,76 В
Метиленовый голубой Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
31
32.
Интервал изменения окраски индикаторов
Ind(ок) + ne Ind(вос)
0,059
[Ind(ок)]
E = Eo + ——— lg ————
n
[Ind(вос)]
[Ind(ок)]
———— = 10 – наблюдаем окраску окисленной
[Ind(вос)]
формы
E = Eo + 0,059/n lg10 = Eo + 0,059/n
32
33.
[Ind(ок)]
——— = 0,1 – наблюдаем окраску восстановл.
[Ind(вос)]
формы
E = Eo + 0,059/n lg 0,1 = Eo – 0,059/n
E = Eo ± 0,059/n
33
34.
Индикаторы окислительно-восстановительного
титрования
1. Специфические – индикаторы, которые взаимодействуют с одной из форм окислительновосстановительной пары с изменением
окраски (крахмал)
2. Редокс-индикаторы — вещества, которые при
определенном потенциале раствора окисляются или восстанавливаются с изменением
окраски
34
35.
Редокс-индикаторы:
1. Обратимые
E = Eo ± 0,059/n
Дифениламин Ео = 0,76 В
бесцветный – фиолетовый
м.р. в воде, готовят 1% р-р в концентр. H2SO4
Ферроин – комплекс Fe(II) с о-фенантролином
Ео = 1,14 В
[FeL3]2+ – e [FeL3]3+
красная
бледно-голубая
Фенилантраниловая кислота (Ео = 1,08 В) и др.
35
36.
2. Необратимые
Метиловый оранжевый
Метиловый красный,
Нейтральный красный
При окислении необратимо исчезает окраска
раствора
36
37.
Индикаторные ошибки окислительновосстановительного титрования
Индикаторная ошибка рассчитывается по
формуле:
n’(X)
Х = ———— · 100
n(X)
n’(X) – количество неоттитрованного вещества
(или избыточно прибавленного титранта)
n(X) – количество вещества, взятого для
титрования
37
38.
Задача. Рассчитать ошибку титрования железа
сульфата (II) раствором KMnO4 в сернокислой
среде при [H+] = 1 моль/л с индикатором
дифениламином (Ео = 0,76 В).
Потенциал системы в т.э.:
E = (mEoв-ля+nEoок-ля )/(m+n)=(1 0,77+5 1,51)/(1+5)
=1,39 B
38
39.
Таким образом,
Изменение окраски произойдет при 0,76 В
Т.э. наступит при 1,39 В, следовательно,
раствор недотитрован.
Потенциал системы рассчитывался по паре
[Fe3+]/[Fe2+] (Eo = 0,77 В) и закончили
титровать при 0,76 В
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
0,76 = 0,77 + 0,059/1 lg[Fe3+]/[Fe2+]
39
40.
lg[Fe3+]/[Fe2+] = – 0,169
[Fe3+]
0,68
———— = 0,68 = ———
[Fe2+]
1
n’(X)
Х = ———— · 100
n(X)
n’(X) = 1 (количество неоттитрованного в-ва)
n(X) = 1 + 0,68 (количество вещества, взятого
для титрования)
40
41.
Ошибка титрования составит:
1
Х = ———— · 100 = 59,5 %
1 + 0,68
Индикаторная ошибка д.б. не более 0,1 %.
41
42.
Задача. Рассчитать и построить кривую
титрования соли олова (II) раствором соли
железа (III), если Eo (Fe3+/Fe2+) = + 0,77 B и
Eo (Sn4+/Sn2+) = + 0,15 B.
Какой из индикаторов вы выберете?
Дифениламин Ео = 0,76 В
Метиленовый голубой Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
Нейтральный красный Ео = 0,24 В
Рассчитать ошибку титрования с индикатором –
метиленовый голубой
42
43.
Sn2+ + 2Fe3+ Sn4+ + 2Fe2+
В начале титрования и до т.э. в растворе будут:
Sn2+, Sn4+, Fe2+
Потенциал системы будет определяться парой
восстановителя Sn4+/Sn2+,
Величина потенциала вычисляется по
уравнению Нернста:
E = Eo + 0,059/2 lg[Sn4+]/[Sn2+]
43
44.
В точке эквивалентности:
E = (mEoв-ля + nEoок-ля )/(m + n)
E = (2 · 0,15 + 1· 0,77) / 3 = 0,36 B
44
45.
Sn2+ + 2Fe3+ Sn4+ + 2Fe2+
за т. э. в растворе будут:
Fe3+, Sn4+, Fe2+
Потенциал будет определяться парой
окислителя Fe3+/Fe2+
Величина потенциала вычисляется по
уравнению Нернста:
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
45
46.
Доб-но
Fe3+
От-но
в%
[Sn4+]
———
[Sn2+]
[Sn4+]
lg ———
[Sn2+]
E
9
9
9/91 10-1
-1
0,12 В
50
50
50/50 1
0
0,15 В
90
90
90/10 101
1
0,18 В
99
99
99/1 102
2
0,21 В
99,9
99,9
99,9/0,1 103
3
0,24 В
100
0,36 B
46
47.
Доб-но
Fe3+
Изб-к
в%
100,1
0,1
0,1/100 10-3
-3
0,59 B
101
1
1/100=10-2
-2
0,65 B
110
10
10/100=10-1
-1
0,71 B
[Fe3+]
———
[Fe2+]
[Fe3+]
lg ———
[Fe2+]
E
Скачок от 0,24 В до 0,59 В
Метиленовый голубой Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
Нейтральный красный Ео = 0,24 В
47
48.
Изменение окраски произойдет при 0,53 В
Т.э. наступит при 0,36 В, следовательно,
раствор перетитрован.
Потенциал системы рассчитывался по паре
окислителя [Fe3+]/[Fe2+] (Eo = 0,77 В)
Закончили титровать при 0,53 В
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
0,53 = 0,77 + 0,059/1 lg[Fe3+]/[Fe2+]
48
49.
lg[Fe3+]/[Fe2+] = – 6,949
[Fe3+]
1·10–7
———— = 1·10–7 = ———
[Fe2+]
1
1·10–7
Х = ——— · 100 = 1·10–5 %
1
n’(X) = 1·10–7 (количество избыточно прибавленного титранта)
n(X) = 1 + 1·10–7 1 (количество вещества,
взятого для титрования)
49
50.
Дихроматометрия
Титрант – 0,0167 М или 0,1 М (1/6 K2Cr2O7)
или 0,1 н. раствор K2Cr2O7
Стандартный раствор можно приготовить по
точной навеске
Cr2O72 + 14H+ + 6ē 2Cr3+ + 7H2O
fэкв(K2Cr2O7) = 1/6
Mэкв(K2Cr2O7) = M(K2Cr2O7)/6
50
51.
K2Cr2O7 – оранжевая окраска
Cr3+ – зеленоватая окраска, однако
интенсивности цвета не хватает для фиксации
конечной точки титрования
Ind – дифениламин, дифениламинсульфоновая
кислота и др.
51
52.
а(K2Cr2O7)теор=Сэкв(K2Cr2O7)·Mэкв(K2Cr2O7)·V (л)
а(K2Cr2O7)прак
Сэкв(K2Cr2O7)прак = —————————
Mэкв(K2Cr2O7) · V(л)
Сэкв(K2Cr2O7)прак
K = ————————
Сэкв(K2Cr2O7)теор
52
53.
Применение:
прямая дихроматометрия
Железа (II) сульфат:
6FeSO4 + K2Cr2O7 + 7H2SO4 3Fe2(SO4)3 +
Cr2(SO4)3 + K2SO4 + 7H2O
2Fe2+ – 2е Fe23+
Сэкв(K2Cr2O7)·Mэкв(FeSO4)
Т(K2Cr2O7/FeSO4) = ———————————
1000
53
54.
Расчет массы и массовой доли (%)
m(FeSO4) = V(K2Cr2O7)·K·T(K2Cr2O7/FeSO4)
V(K2Cr2O7)·K·T(K2Cr2O7/FeSO4)·100
(FeSO4) = ——————————————, %
а(FeSO4)
54
55.
Калия иодид:
6KI + K2Cr2O7 + 7H2SO4 3I2 + Cr2(SO4)3 +
4K2SO4 + 7H2O
2I– – 2е I2
Сэкв(K2Cr2O7)·Mэкв(KI)
Т(K2Cr2O7/KI) = ——————————
1000
55
56.
Кислота аскорбиновая:
O O
OH OH
H
3
O
+ K2CrO4 + 4H SO
2
4
O
H
CHOH
O
CHOH
CH2OH
CH2OH
C21+ – 2е
+ Cr2(SO4)3 + K2SO4 + 7H2O
O
C22+
С(1/6K2Cr2O7)·M(1/2Аск.к.)
Т(K2Cr2O7/Аск.к.) = ———————————
1000
56
57.
Обратная дихроматометрия применяется для
определения спирта этилового:
3CH3CH2OH + 2K2Cr2O7 + 8H2SO4
избыток
3CH3COOH + 2Cr2(SO4)3 + 2K2SO4 + 11H2O
K2Cr2O7 + 6KI + 7H2SO4 3I2 + Cr2(SO4)3 +
остаток
4K2SO4 + 7H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
57
58.
Расчет массовой доли (%)
(V(K2Cr2O7) K V(Na2S2O3) K) Т(Na2S2O3/Сп) 100
(Сп) = (%)
а (CH3CH2OH)
Сэкв(Na2S2O3) Мэкв(Спитра)
Т(Na2S2O3/Сп) =
1000
-1
CH3—CH2OH – 4 е
+3
CH3—COOH
58
59.
Преимущества дихроматометрии перед
перманганатометрией
1. Титрант можно приготовить по точной
навеске
2. Титрование можно проводить в присутствии
HCl
Eo(Cr2O72–/2Cr3+) = 1,33 B
Eo(Cl2/2Cl–) = 1,33 B
При титровании Cl– не окисляются Cr2O72–ионами
59
Индикаторная ошибка — титрование
Cтраница 2
Чтобы окраска окислительно-восстановительного индикатора изменялась при титровании резко и индикаторная ошибка титрования была незначительной, интервал перехода индикатора должен находиться в пределах скачка потенциалов на кривой титрования.
[16]
Чтобы окраска окислительно-восстановительного индикатора изменялась при титровании резко и индикаторная ошибка титрования была незначительной, необходимо, чтобы область перехода индикатора находилась в пределах скачка потенциалов на кривой титрования.
[17]
Поэтому даже при правильном выборе индикатора допускается погрешность, называемая индикаторной ошибкой титрования. Неправильный выбор индикатора может совершенно исказить результаты анализа. Рассмотрим теорию индикаторов и правила их выбора при различных случаях титрования.
[19]
Кроме метода кривых титрования, для выбора индикатора применяют способ вычисления индикаторной ошибки титрования.
[21]
В методе кулонометрического титрования имеется пять возможных источников ошибок: 1) изменение силы тока в процессе электролиза, 2) отклонение течения процесса от 100 % — ного выхода по току, 3) ошибки в измерении силы тока, 4) ошибки в измерении времени, 5) индикаторная ошибка титрования, обусловленная несовпадением точки эквивалентности и конечной точки титрования. Последнее характерно для всех титриметрических методов. Если индикаторная ошибка является доминирующим фактором, то оба метода сравнимы по надежности получаемых результатов.
[22]
Действительно из табл. 11 видно, что рН 4 получается в тот момент, когда при бавлено 99 9 мл раствора NaOH. Следовательно, индикаторная ошибка титрования составляет здесь 0 1 мл на 100 мл, а при титровании 25 мл-всего 0 025 мл.
[23]
Выше было показано, что рТ индикатора, отвечающий точке конца титрова-ия, как правило, не совпадает с рН раствора в точке эквивалентности. Это вызывает индикаторную ошибку титрования. Вследствие несовпадения рТ выбранного индикатора и рН титруемого раствора в точке эквивалентности раствор обычно либо несколько перетитровывают, либо, наоборот, недотитровывают. В результате по окончании титрования раствор содержит некоторый избыток свободной кислоты или свободной щелочи. Если значение рТ меньше чем рН в точке эквивалентности, то ошибка вызывается избытком Н — иона и называется водородной ошибкой Н — ошибкой. Если, наоборот, рТ больше, чем в точке эквивалентности, то ошибка вызвана избытком ОН — — ионов и называется гидр-оксильной ошибкой ОН — — ошибкой. Если титруют не сильные, а слабые кислоты и основания, когда кислота или основание присутствуют практически в неионизированной форме, то говорят о кислотной ошибке, или HAn-ошибке, соответственно — об основной ошибке, или МеОН — ошибке. При титровании щелочей или слабых оснований знаки у Н — ошибки ( и НАп-ошибки) ОН — — ошибки ( МеОН — ошибки), естественно, меняются на обратные.
[24]
Она называется индикаторной ошибкой титрования.
[25]
Наряду с построением кривых титрования существует также другой способ выбора индикатора ( стр. Напомним, что индикаторная ошибка титрования представляет собой ту погрешность, которая обусловлена несовпадением показателя титрования примененного индикатора с величиной рН в точке эквивалентности. Вследствие этого несовпадения раствор обычно либо несколько пере-гитровывают, либо, наоборот, недотитровывают. В результате по окончании титрования раствор содержит некоторый избыток свободной кислоты или свободной щелочи.
[26]
Наряду с построением кривых титрования существует также другой способ выбора индикатора ( стр. Напомним, что индикаторная ошибка титрования представляет собой ту погрешность, которая обусловлена несовпадением показателя титрования примененного индикатора с величиной рН в точке эквивалентности. Вследствие этого несовпадения раствор обычно либо несколько пере-титровывают, либо, наоборот, недотитровывают. В результате по окончании титрования раствор содержит некоторый избыток свободной кислоты или свободной щелочи.
[27]
Наряду с построением кривых титрования существует также другой способ выбора индикатора ( стр. Напомним, что индикаторная ошибка титрования представляет собой ту погрешность, которая обусловлена несовпадением показателя титрования примененного индикатора с величиной рН в точке эквивалентности. Вследствие этого несовпадения раствор обычно либо несколько пере-гитровывают, либо, наоборот, недотитровывают. В результате по окончании титрования раствор содержит некоторый избыток свободной кислоты или свободной щелочи.
[28]
Окраска индикаторов метода кислотно-основного титрования меняется в определенном интервале значений рН часто не строго в точке эквивалентности, а с некоторыми отклонениями как в ту, так и в другую сторону. Эту погрешность называют индикаторной ошибкой титрования.
[29]
Казалось бы, что такие значительные отклонения от точки эквивалентности должны обусловить значительную индикаторную ошибку титрования ( стр. В данном случае все четыре наиболее часто применяемых индикатора дают практически совпадающие результаты и могут быть с успехом применены для титрования.
[30]
Страницы:
1
2
3
Построение кривой титрования по методам окислительно-восстановительных реакций и нейтрализации
Федеральное Агентство Образования
АСТРАХАНСКИЙ ГОСУДАРСТВЕННЫЙ
УНИВЕРСИТЕТ
Курсовая работа
по аналитической химии на тему:
«Построение кривой титрования по
методам окислительно-восстановительных реакций и нейтрализации
Астрахань-2011 год
Оглавление
1. Введение
2.Сущность и понятия титраметрического анализа
2.1 Окислительно-восстановительное титрование
2.2 Условия проведения реакций окислительно-восстановительного
титрования
2.3 Виды окислительно-восстановительного титрования
2.4 Индикаторы метода
3. Пермангонатометрия
3.1 Сущность метода
3.2 Условия проведения
3.3 Индикаторы
4. Расчет точек кривой титрования, потенциалов, построение кривой
титрования
4.1 Окислительно-восстановительное титрование 0,3 М HBr — 0,1М KMnO4
4.2 Расчет точек кривой титрования, потенциалов
4.3 Построение кривой титрования
4.4 Подборка индикатора. Расчет индикаторных ошибок титрования
4.5 Вывод
5. Кислотно-основное титрование. Основные понятия
5.1 Индикаторы
5.2 Классификация индикаторов
5.3 Окраска индикаторов
5.4 Интервал перехода индикаторов
5.5 Исследование индикаторов
5.6 Выбор индикатора
5.7 Влияние растворителей
5.8 Влияние температуры
5.9 Индикаторная ошибка
5.10 Типы индикаторных ошибок
6. Кислотно-основное титрование 0,1 M KCN — 0,3 M H2SO4
6.1 Расчет точек кривой титрования
6.2 Построение кривой титрования
6.3 Подборка индикатора. Расчет индикаторных ошибок титрования
7. Вывод
8. Список литературы
1. Введение
Одним из самых старых и даже в настоящее время
распространенных методов химического анализа является титраметрический анализ.
Свое применение находят множество видов титраметрического анализа, такие как
кислотно-основное, окислительно-восстановительное, осадительное,
комплексонометрическое, титрование неводных растворов, физико-инструментальные
методы титрования. Используя результаты титрования, можно без труда определить
различные вещества.
Для проведения анализа нужно знать некоторые понятия и
закономерности в том числе и конечную точку титрования. Для ее определения
часто используются индикаторы. Для подбора индикатора необходимо, сделав
теоретические расчеты, построить кривую титрования, определить скачок
титрования и рассчитать индикаторные ошибки титрования.
Поэтому целью данной работы является выбор индикатора и
построение кривой титрования. Задачами работы являются расчет точек титрования,
выявление области скачка титрования, вычисление точки эквивалентности,
вычисление индикаторных ошибок титрования.
титрование окислительный восстановительный реакция
2.Сущность
и понятия титраметрического анализа
) Титраметрический, или объемный метод, анализ — метод
количественного анализа, основанный на измерении объема (или массы) реагента,
затраченного на реакцию с определяемым веществом. Другими словами,
титраметрический анализ — это анализ, основанный на титровании.
Титрование — процесс определение вещества постепенным
прибавлением небольших количеств титранта, при котором каким-либо способом
обеспечивается обнаружение точки, когда определяемое вещество полностью
прореагировало.
Титрант — раствор, содержащий активный реагент Т, с помощью
которого проводят титрование.
Аликвотная доля (аликвота) — это точно известная часть
анализируемого раствора, взятая на анализ.
Точка эквивалентности (Т. Э.) — такая точка титрования, в
котором количество прибавленного титранта Т эквивалентно количеству титруемого
вещества.
Конечная точка титрования (КТТ) — точка титрования, в
которой некоторое свойство раствора оказывает резкое и заметное изменение. КТТ
чаще всего не совпадает с точкой эквивалентности.
Интервал перехода индикатора — область концентрации
ионов H+, Men+, или других ионов, в пределах которых глаз
способен обнаружить изменение в оттенке, интенсивности в окраске, флуоресценции
или другого способа визуального индикатора, вызванного изменением соотношения
двух соответствующих форм индикатора.
Степень оттитрованности (f) — отношение объема V (T) добавленного титранта к
объему V
(TЭ) титранта,
соответствующему точке эквивалентности:
f=V (T) /V (TЭ).
Кривая титрования — графическое
изображение зависимости изменения концентрации С (х) определяемого вещества х
или некоторого связанного с ним свойства системы (раствора) от объема V (T) прибавленного титранта
Т. Величина С (х) в ходе титрования изменяется на несколько порядков, поэтому
кривая титрования часто строится в координатах lgС (х) — V (T).
Различают линейные, логарифмические, дифференциальные,
билогарифмические кривые титрования.
Классификация методов титраметрического анализа.
1. Кислотно — основное титрование — основано на реакции
переноса протонов от одной реагирующей единицы к другой в растворе. Различат
ацидиметрию и алкалиметрию.
. Окислительно-восстановтельное титрование — сопровождается
переходом 1 или большого числа электронов от иона — донора или молекулы
(восстановителя) к акцептору (окислителю).
. Осадительное титрование — титруемое вещество при
взаимодействии с титрантом выделяется из раствора в виде осадка.
. Комплексонометрическое титрование — титрование вещества
раствором такого соединения, которое образует с титруемым веществом слабодиссоциирующий
растворимый комплекс.
2.1
Окислительно-восстановительное титрование
Классификация редокс-методов
Известно несколько десятков различных методов 0В титрования.
Обычно их классифицируют следующим образом.
Классификация по характеру титранта. В этом случае методы ОВ
титрования подразделяют на две группы:
оксидиметрия — методы определения восстановителей с
применением титранта-окислителя;
редууктометрия — методы определения окислителей с применением
титранта-восстановителя.
Классификация по природе реагента, взаимодействующего с
определяемым веществом. Ниже после названия соответствующего метода в скобках
указано основное действующее вещество этого метода: броматометрия (KBrO3), бромометрия (Br2), дихроматометрия (K2Cr2O7), иодатометрия (KIO3), иодометрия (I2), нитритометрия (NaNO2), пермангонатометрия (KMnO4), цериметрия (Ce (SO4) 2)),
титанометрия (Ti3+), ванадатометрия (NH4VO3) и др. Реже применяются некоторые другие методы
0В титрования, такие, аскорбинометрия (аскорбиновая кислота).
2.2
Условия проведения реакций окислительно-восстановительного титрования
Условия, применяемые в методах
окислительно-восстановительного титрования должны отвечать ряду требований,
важнейшими из которых являются следующие:
Реакции должны протекать практически до конца.
Реакция должна протекать стехиометрически, побочные эффекты должны
быть исключены.
Конечная точка титрования должна определятся точно и
однозначно либо с индикатором или нет.
2.3
Виды окислительно-восстановительного титрования
1) Прямое окислительно-восстановительное титрование проводят
тогда, когда окислительно-восстановительная реакция удовлетворяет основным
требованиям к реакциям этого вида титрования. Определяемое вещество
непосредственно взаимодействует с титрантом.
) Обратное окислительно-восстановительное титрование проводят
тогда, когда применение прямого титрования нецелесообразно по тем или иным
причинам. К аликвоте анализируемого раствора, содержащего определяемый
компонент х, прибавляют точно известное избыточное количество вещества А.
Выдерживают раствор некоторое время для обеспечения полноты реакции между х и
А. Непрореагирующий избыток вещества А оттитровывают стандартным раствором
титранта Т.
) Заместительное окислительно-восстановительное титрование
применяют для определения веществ как вступающих, так и не вступающих в
окислительно-восстановительные реакции. К аликвоте анализируемого раствора,
содержащего определяемый компонент, прибавляют избыточное количество реагента.
Выделившееся вещество оттитровывают определенным титрантом.
2.4
Индикаторы метода
В титрометрическом физическом методе определяют КТТ
индикаторным методом. При этом индикатор может играть роль либо реагента,
участвующего в окислительно-восстановительной реакции, либо являться
специальным вводимым индикатором. В соответствии с этим индикаторы в
окислительно-восстановительном титровании можно классифицировать следующим
образом:
). Индикатор — реагент, участвующий в
окислительно-восстановительной реакции (пермангонатометрия)
). Индикатор — вещество, вступающий в непосредственное
взаимодействие с окислителем или восстановителем (участвующими в
окислительно-восстановительной реакции) с образованием различных соединений
(крахмал).
). Индикатор — вещество, которое при определенном потенциале
раствора окисляется или восстанавливается с изменением окраски
(окислительно-восстановительные индикаторы).
Окислительно-восстановительные индикаторы бывают обратимыми и
необратимыми. Обратимые индикаторы меняют окраску обратимо при потенциале
раствора в точке эквивалентности или вблизи ее и при этом не разрушаются.
Необратимые индикаторы изменяют окраску при достижении определенного значения
потенциала в точке эквивалентности или вблизи ее и при этом необратимо
разрушаются. Пример — метиловый оранжевый, метиловый красный, нейтральный
красный.
3.
Пермангонатометрия
3.1
Сущность метода
Пермангонатометрический метод анализа основан на реакции
перманганата калия с восстановителями, преимущественно в кислой среде по схеме:
MnO4 — +8H+ +5e↔Mn2++4H2O,
f=1/2=1/5.
В анализе некоторых органических соединений используется
восстановление в сильно щелочной среде по уравнению:
MnO4 — +e↔ MnO42-, f=1
Стандартный 0В потенциал редокс-пары МnО4/ Мn2+ имеет довольно высокое
значение и при комнатной температуре равен 1,51 В. Поэтому кислым раствором
перманганата калия можно оттитровать целый ряд восстановителей, причем
большинство таких 0В реакций протекает с высокой скоростью. С ростом
концентрации ионов водорода в растворе реальный потенциал рассматриваемой
редокс-пары возрастает и эффективность перманганат-иона как окислителя
повышается.
Е = Е° + (0,059/5) 1g ([МnО4—,] [}H+] 5/[Мn2+])
Поскольку в 0В полуреакции участвуют 5 электронов, то
молярная масса эквивалента перманганата калия как окислителя в кислой среде
равна
М (1/5КМnО4) = М (КМ04) /5 = 31,608 г/моль,
В нейтральной среде перманганат-ион восстанавливается до
диоксида марганца МnО2:
МnО4—, + Зе + 2Н20 = МnО2 + 40Н—
Стандартный 0В потенциал редокс-пары МnО4—|
МnО2,
сравнительно невелик и при комнатной температуре равен Д° = 0,60 В, поэтому
нейтральной среде эффективность перманганата калия как окислителя значительно
ниже, чем в сильнокислых растворах. Кроме того, образующийся в результате 0В
реакции бурый осадок диоксида марганца затрудняет фиксацию КТТ, обладает
развитой поверхностью и может адсорбировать определяемое вещество, что
увеличивает ошибку анализа. Поэтому как титрант-окислитель перманганат калия в
нейтральной среде практически не применяется.
В сильнощелочных средах перманганат-ион восстанавливается до
манганат-иона МпО42-:
МnО4—+е=МnО42-
Образующийся манганат-ион обладает зеленой окраской умеренной
интенсивности, окрашивает раствор в зеленый цвет, что затрудняет обнаружение
изменения окраски раствора и фиксацию КТТ.
Манганат-ион вступает в реакцию с водой:
МnО42 — + 2Н20 = 2Мn04 + МnО2 ↓
+40Н-
с образованием бурого осадка диоксида марганца и
перманганат-ион что искажает результаты анализа.
Стандартный 0В потенциал редокс-пары МnО4—|
МnО42 — невелик
при комнатной температуре равен Е° = 0,56 В, т.е. в щелочных средах перманганат
калия как окислитель менее эффективен, чем в кислых растворах. Кроме того,
щелочные растворы способны поглощать диоксид углерода из воздуха с образованием
гидроксокарбонатных соединений, что также затрудняет проведение количественного
анализа.
С учетом всех этих обстоятельств перманганат калия как
титрант-окислитель в щелочных средах практически не применяется.
Таким образом, на основе вышеизложенного можно сделать вывод
о том, что перманганатометрическое титрование целесообразно проводить в
сильнокислых средах.
3.2
Условия проведения
При проведении перманганатометрического титрования необходимо
соблюдать, по крайней мере, следующие основные условия.
) Влияние рН среды. Перманганатометрическое титрование
проводят в сильно кислой среде при концентрации ионов водорода [НзО+]
=1-2 моль/л. Кислая среда создается введением серной кислоты. Азотную кислоту
применять нельзя, так как она сама является сильным окислителем и может
окислять определяемое вещество. Хлороводородную кислоту в перманганатометрии
также не используют, так как хлорид-ионы окисляются перманганат-ионами до хлора
по схеме:
Cl — +2МnО4 — +16Н+ =5С12
+2Мn2+ +8Н20
При этом часть титранта расходуется на окисление
хлорид-ионов, что вызывает перерасход титранта и увеличивает ошибку анализа. В
обычных условиях эта реакция идет медленно, однако ускоряется в присутствии
соединений железа.
В сернокислой среде указанные побочные процессы отсутствуют,
поэтому перманганатометрическое титрование ведут в сернокислой среде.
) Влияние температуры. Чаще всего перманганатометрическое
определение проводят при комнатной температуре. Исключением является реакция
перманганат-иона с щавелевой кислотой и оксалатами, которую проводят при
нагревании титруемого раствора.
) Фиксация конечной точки титрования. При
перманганатометрическом титровании обычно не применяют посторонний индикатор,
так как сам титрант — раствор перманганата калия — обладает интенсивной
малиново-фиолетовой окраской. Прибавление одной избыточной капли титранта в ТЭ
приводит к окрашиванию титруемого раствора в розовый цвет. Так, чтобы придать
отчетливую окраску 100 мл воды достаточно прибавить всего 0,2 мл раствора
перманганата калия с молярной концентрацией эквивалента 0,1 моль/л. Окраска
раствора в КТТ неустойчива, раствор постепенно обесцвечивается. Это происходит
вследствие того, что избыточные перманганат-ионы, придающие раствору розовую
окраску, взаимодействуют с образовавшимися катионами марганца (2) Мn2+:
МnО4—+ЗМn2++Н20=5МnО2↓+4Н+
Хотя повышенная кислотность раствора способствует смещению
равновесия влево, однако константа равновесия этой реакции велика и составляет
~1047. Поэтому реакция протекает даже в кислой среде. Правда,
скорость данной реакции в обычных условиях мала, поэтому окраска раствора в КТТ
ослабляется медленно, в течение одной-двух минут. Тем не менее, указанное
обстоятельство необходимо иметь в виду во избежание перетитровывания раствора.
В некоторых (редких) случаях перманганатометрическое
титрование ведут очень разбавленным раствором титранта. В такой ситуации для
более четкого фиксирования КТТ в титруемый раствор вводят редокс-индикатор, например
ферроин или N-фенилантраниловую
кислоту.
) Ход титрования. Обычно в перманганатометрии к раствору
определяемого вещества медленно, по каплям прибавляют раствор титранта, для
того чтобы в растворе не было локального избытка окислителя — перманганат-иона
и не протекали бы побочные процессы.
Для титрования применяют бюретки со стеклянными кранами;
использование резиновых трубок исключается, так как резина взаимодействует с
перманганатом калия.
3.3
Индикаторы
В пермангонатометрии часто обходятся без применения
специального индикатора, так как перманганат имеет интенсивную окраску, и
избыточная капля реагента легко обнаруживается по собственной окраске.
Титрование раствора до бледно-розового окрашивания, не исчезающего в течение 30
секунд, — обычный способ фиксирования точки эквивалентности в методе. При
титровании с разбавленными растворами применяют резонс-индикаторы, такие как
дифениламинсульфокислота или ферроин.
4.
Расчет точек кривой титрования, потенциалов, построение кривой титрования
4.1
Окислительно-восстановительное титрование 0,3 М HBr — 0,1М KMnO4
16HBr + 2KMnO4=2KBr+2MnBr2+5Br2+8H2O+7+5ё→Mn2+ окислитель
Br—-2ё→Br20 восстановитель
Концентрация HBr =0.3 M, концентрация KMnO4=0.1 M.
Сн (HBr) = См/ fэкв-ти = 0.3/1 = 0.3
Сн (KMnO4) = См/ fэкв-ти = 0.1*5 = 0.5
φ0 (MnO4—/ Mn2+) =1.51 В, φ0 (Br—/ Br20) =1.09 В
Пусть объем раствора KMnO4 будет равен 10мл.
С (HBr) *V (HBr) = С (KMnO4) *V (KMnO4)
V (KMnO4) = С (HBr) *V (HBr) / С (KMnO4) = 0.3*10/0,5 = 6 мл.
4.2
Расчет точек кривой титрования, потенциалов
До точки эквивалентности считаем по титруемому веществу:
φ= φ0+0.059/n*lg ([HBr—] 2/
([Br2—])
1.1% добавили от необходимого объема
6 мл — 100%, Х мл — 1%
Х=0,06 (мл) — добавленный объем KMnO4
Найдем Сн (KMnO4) прореагировавшего с
учетом разбавления:
Сн (KMnO4) = Сн (KMnO4) нач. *Vдоб/Vобщ. = 0.06*0.5/ (10+0.06)
=0.002982
По закону эквивалентов:
Сн (Br—) прореаг. = Сн (MnO4—) прореаг=0.002982
Найдем Сисх. (Br—) с учетом разбавления:
Сисх. (Br—) = Сн (Br—) *Vнач. (Br—) /Vобщ. = 0.3*10/ (10+0.06)
=0.2982
Найдем оставшуюся
С (Br—): Соств. =Снач. — Спрореаг.
= 0.2982 — 0.002982= 0.2952
φ= 1.09 + 0.059/1*lg (0.0029822/
0.2982) = 0.8900 B
.25% добавили от необходимого объема
мл — 100%, Х мл — 25%
Х=1.5 (мл) — добавленный объем KMnO4
Найдем Сн (KMnO4) прореагировавшего с
учетом разбавления:
По закону эквивалентов:
Сн (Br—) прореаг. = Сн (KMnO4) прореаг =
0.06521
Найдем Сисх (Br—) с учетом разбавления:
Сисх. (Br—) = Сн (Br—) *Vнач. (Br—) /Vобщ. = 0.02*10/ (10+1.5) =
0.26083
Найдем оставшуюся С (Br—):
Соств. = Снач. — Спрореаг. =
0.26083 — 0.06521= 0.19559
φ= 1.09 + 0.059/1*lg (0.065212/0.26083)
= 0.8911 B
.50% добавили от необходимого объема
6 мл — 100%
Х мл — 50%
Х=3 (мл) — добавленный объем KMnO4
Найдем Сн (KMnO4) прореагировавшего с
учетом разбавления:
Сн (KMnO4) = Сн (KMnO4) нач. *Vдоб/Vобщ. = 3*0.5/ (10+3) = 0.1153
По закону эквивалентов:
Сн (Br—) прореаг. = Сн (KMnO4) прореаг=0.1153
Найдем Сисх. (Br—) с учетом разбавления:
Сисх. (Br—) = Сн (Br—) *Vнач. (Br—) /Vобщ. = 0.3*10/ (10+3) = 0.2307
Найдем оставшуюся
С (Br—): Соств. = Снач. — Спрореаг
= 0.2307
.1153= 0.1154
φ= 1.09 +0.059/1*lg (0.11532/0.2307)
= 9788 B
.99.9% добавили от необходимого объема
6 мл — 100%
Х мл — 99.9%
Х = 5.994 (мл) — добавленный объем KMnO4
Найдем Сн (KMnO4) прореагировавшего с
учетом разбавления:
Сн (KMnO4) = Сн (KMnO4) нач. *Vдоб/Vобщ. = 5.994 *0.15/ (10+5.994)
= 0.1873
По закону эквивалентов:
Сн (Br—) прореаг. = Сн (KMnO4) прореаг=
0.1873
Найдем Сисх. (Br—) с учетом разбавления:
Сисх. (Br—) = Сн (Br—) *Vнач. (Br—) /Vобщ. = 0.3*10/ (10+5.994)
=0.1875
Найдем оставшуюся
С (Br—): Соств. =Снач. — Спрореаг.
= 0.1875-0.1873= 0.0002
φ= 1.09+0.059/*lg (0.18732/0.1875)
= 1.00416 B
.100% добавили от необходимого объема. Точка
эквивалентности
. φ= (φок*nок+ φвос*nвос) / (nок+ nвос) = (1.51*5+1.09*1) /
(1+5) = 1,44
. lgKp= (φок — φвос) *n/0.059= (1.51-1,09) *6/0.059 = 2.7966
Kp=102.7966, Kp=1/ [HBr—] 2* [MnO4—] 6
Пусть [MnO4] = х моль/литр, тогда [HBr—] =х. [HBr—] с учетом разбавления
равна:
[HBr—] = Снач. *Vнач. /Vобщ. =0.06*10/14 = 0.042857
(общая концентрация.)
Тогда [HBr—], оставшегося в растворе равна (0.007-х).
С (KMnO4) = Сн (KMnO4) нач. *Vдоб/Vобщ. = 0.15*4/14 = 0.007
Уточним концентрацию [Mn2+] в точке эквивалентности
с учетом разбавления:
[Mn2+] = 0.007-х
Константа равновесия с учетом следующих соотношений принимает
вид:
101.72=1/x8
10101.72= x-8
√10101.72=х-8
х=7*10-16.53 — равновесная концентрация [MnO4—]
φ=
1.53+0.058/5*lg ([MnO4—] 2/ ([Mn2+])
= 1.53+0.059/5*lg ([7*10-16.53] / ([0.007]) =1.36
6.110% добавили от необходимого объема, раствор
перетитрован
6 мл — 100%
Х мл — 10%
Х=0.6 (мл) — избыточный объем KMnO4
φ= φ 0+0.059/1*lg ([MnO4—] 2/ ([Mn2+])
Найдем избыточную См (KMnO4) с учетом разбавления:
См. (KMnO4) изб. = См (KMnO4) нач. *Vдоб/Vобщ. =0.6*0.5/ (10.06) =0.0036
Сколько молей Mn2+ получили, столько в растворе и осталось:
[Mn2+] оств. = Снач. *Vдоб/Vобщ. = 0.15*3.3/ (10.06)
=0.0363
φ= 1.53+0.059/5*lg ([0.0036] / ([0.0363])
=1.51
4.3
Построение кривой титрования
Таблица1. Точки для построения кривой титрования по методу
ОВР
|
Процент |
Потенциал, В |
|
1% добавили от |
0.8900 |
|
25% добавили от |
0.8911 |
|
50% добавили от |
0.9788 |
|
99.9% добавили |
|
|
100% добавили |
1.36 |
|
110% добавили |
1.51 |
График
1. Кривая титрования 0,3 М HBr — 0,1М KMnO4
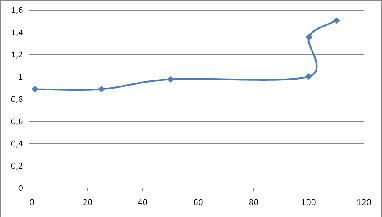
4.4 Подборка индикатора. Расчет индикаторных ошибок титрования
1. Пусть φ (2,2-дипиридин) =1.33<φ в точке эквивалентности, равного 1.36. Следовательно раствор HBr недотитрован.
φ= φ 0+0.059/2*lg ([HBr] 2/ ([Br—])
Пусть [HBr)] = (100-х) 2, а [Br—)] =х.
.33=0.94+0.058/2*lg ( (100-х) 2/х)
.058/2*lg ( (100-х) 2/х) =0.39
lg ( (100-х) 2/х) =13.44
(100-х) 2/х=1013,448
(100-х) 2=1013.44**х
-200х=1013.44**х2
х=3.56*10-12%
. Пусть φind=1.4<φ в точке эквивалентности, равного1.36. Следовательно раствор HBr перетитрован раствором KMnO4.
.4= 1.53+0.058/5*lg ([MnO4—] / ([Mn2+])
Пусть [MnO4—] =х, тогда [Mn2+] =100
.4= 1.53+0.058/5*lg ([х] / ([100])
.66= 0.0116*lg ([х] / [100])
х/100=3.16*10-13
х=3.16*10-15%
4.5
Вывод
С помощью расчетов точек кривой титрования по методу ОВР
кривой 0,3 М HBr — 0,1М KMnO4, вычислили скачок титрования, равный от 0.99
(99.9%) до 1.51 (110%). Так как скачок небольшой, построили кривую титрования.
С помощью расчета индикаторных ошибок, подобрали оптимальные индикаторы. С
индикатором 2,2-дипиридином индикаторная ошибка составила большая индикаторная
ошибка. Со вторым индикатором ошибка составила 3.16*10-15%.
Следовательно, титровать раствор HBr раствором KMnO4 можно со вторым
индикатором.
5.
Кислотно-основное титрование. Основные понятия
Кислотно-основное титрование — это метод определения
кислот, оснований, солей, основанный без реакции взаимодействия между кислотой
и основанием.
В общем виде можно записать:
НА + В = А — + НВ+
В водных растворах — это реакция нейтрализации:
Н30+ + ОН =2Н2О
Поэтому метод кислотно-основного титрования также называют
методом нейтрализации
Реагент обычно применяют в виде раствора точно известной
концентрации. Такой раствор называют типрованным, титрантом или
рабочим раствором. Рабочий раствор постепенно прибавляют к раствору
определяемого вещества до тех пор, пока не будет достигнуто эквивалентное
отношение реагирующих вещества. Измеряют объём рабочего раствора,
израсходованного на титрование, и вычисляют количество определяемого вещества.
При титровании весьма важно определить точку эквивалентности, т е момент
титрования, когда достигнуто эквивалентное отношение реагирующих компонентов —
определяемого вещества и реагента.
Рассмотрим случай, когда b молей вещества X реагирует
с а молями реагента R, т.е. реакция протекает в соответствии с уравнением:
bX+aR↔XbRa
В любой момент титрования концентрация X представляет собой
некоторую конечную величину, отличную от нуля, т.к. продукт реакции XR способен в той или иной
степени, в результате протекания обратной реакции, давать снова исходный
вещества X и R
в точке эквивалентности концентрации реагирующих веществ становятся
эквивалентными. Для записанной выше реакции это означает, что
a [X] =B [P].
Точку эквивалентности определяют экспериментально посредством
индикаторов.
При использовании любого способа определения точки
эквивалентности необходимо иметь чёткое представление об изменении концентрации
реагирующих веществ в процессе титрования. Это изменение обычно выражают кривой
титрования. Кривая титрования — графическое отражение зависимости концентрации
определяемого вещества от количества прибавленного реагента. Обычно, кривая
титрования передаёт зависимость логарифма концентрации определяемого вещества.
взятого с обратным знаком (-lgX), от объёма прибавленного рабочего раствора
известной концентрации.
5.1
Индикаторы
Существую различные методы установления точки
эквивалентности. Их можно разделить на 2 группы:
первая группа — индикаторные методы вторая группа —
физико-химические методы. Ко второй группе относятся потенциометрический метод,
амперометрический метод, метод радиоактивных индикаторов и др.
Индикатор — это вещество, которое претерпевает химическое
превращение, сопровождающееся внешним эффектом, при концентрациях реагента или
определяемого вещества. характерных для точки эквивалентности.
Индикатор — это вещество, которое проявляет видимое
изменение в точке эквивалентности или вблизи неё.
Индикатор — вещество в присутствии, которого в растворе
становятся видимым конец титрования. Как видно, можно давать различные
определения понятию индикатор. Из приведенных выше наиболее подходящим является
определение 2, которое отражает факт изменения индикатора не только в точке
эквивалентности, но и вблизи нее, а также является наиболее кратким.
Видимый эффект может состоять в изменении, возникновении или
исчезновении окраски, образовании или растворении малорастворимого соединения и
лр Существенно при этом, чтобы внесший эффект проявлялся только при такой
концентрации определяемого вещества X или реагента R, которая соответствует
точке эквивалентности т.е. индикатор должен реагировать с реагентом или с
определяемым веществом только при такой концентрации одного из них, какая
создаётся в точке эквивалентности.
Момент титрования, когда наблюдается внешний эффект реакции
между индикатором и рабочим раствором реагента R (или определяемым
веществом X) называют конечной точкой титрования. Расхождение между
конечной точкой титрования и точкой эквивалентности, т е неравенство рRэкв и рТ (или рХэкв и рТ)
является одной из причин возникновения погрешности титрования.
5.2
Классификация индикаторов
В основе классификации индикаторов могут быть различные
признаки. По технике применения различают внутренние и внешние
индикаторы.
Внутренние индикаторы — вещества, которые
вводят непосредственно в титруемый раствор. Конечную точку титрования
устанавливают по изменению цвета индикатора, по появлению или исчезновению
осадка или по какому-либо другому внешнему эффекту реакции. Внутренние
индикаторы широко распространены. Внешние индикаторы применяют в случаях, когда
использование внутренних индикаторов невозможно. Например, ионы никеля можно
титровать раствором диметилглиоксима Однако красный цвет осадка
диметилглиоксимата никеля мешает наблюдению эффекта реакции, вызванного
прибавлением индикатора. Поэтому в процессе титрования время от времени
отбирают каплю раствора, переносят её на часовое стекло и прибавляют реагент
(тот же диметилглиоксим) на ионы никеля. Титрование ведут до отрицательной
реакции на определяемый ион. Применение внешних индикаторов требует затраты
дополнительного времени и высокой квалификации аналитика.
По обратимости внешнего эффекта реакции различают
обратимые и необратимы индикаторы. Обратимые индикаторы — это
соединения, способные существовать в двух (или более) формах, причём переход
одной формы в другую обратим. К этому типу принадлежит большинство известных
индикаторов. Так, обратимые окрашенные индикаторы легко изменяют окраску в
зависимости от избытка или недостатка введённого реагента или определяемого
вещества. Примером может служить метиловый оранжевый для которого переход
красной формы индикатора в жёлтую и наоборот может происходить много раз в
зависимости от изменения концентрации ионов водорода. Обратимые индикаторы удобны
тем, что при случайном введении слишком большого количества рабочего раствора
определение всё же можно провести правильно. Для этого к перетитрованному
раствору следует добавить ещё некоторое количество определяемого вещества (при
этом восстанавливается первоначальная окраска индикатора) и осторожно
дотировать раствор.
Необратимые индикаторы — это соединения,
которые разрушаются при введении избытка реагента и цвет которых не
восстанавливается от дополнительного прибавления анализируемого раствора. Тот
же метиловый оранжевый может быть примером необратимого индикатора в реакциях
окисления-восстановления. Трёхвалентную сурьму титруют раствором бромата калия
с метиловым оранжевым. До тех пор, пока в растворе нет избытка окислителя,
индикатор окрашен в красный цвет. После точки эквивалентности некоторое
количество раствора бромата калия приводят к окислению индикатора, вследствие
чего раствор обесцвечивается. Естественно, что после прибавления раствора
трёхвалентной сурьмы окраска не восстановится, так как весь индикатор разрушен.
Необратимые индикаторы менее удобны и применяются редко.
Индикаторы различают по типу химической реакции, в которой их
применяют. Например, кислотно-основные индикаторы, индикаторы осадительного
титрования и т.д. для титрования мутных и окрашенных растворов применяют
люминесцентные и хемолюминесцентные индикаторы. Использование люминесцентных
индикаторов основано на применении веществ, которые при освещении у. ф. лучами
изменяют характер свечения в зависимости от изменения свойств среды (рН среды,
окислительно-восстановительного потенциала и т. д). Поэтому люминесцентные
индикаторы применяют в методах кислотно-основного титрования,
комплексообразования и окисления-восстановления.
В зависимости от химического строения индикатора, можно выделить
следующие группы
. Азоиндикаторы.
. Нитроиндикаторы.
. Фталеины.
. Сульфофталенны и др.
Существует несколько теорий строения кислотно-основных
индикаторов. Наиболее известными являются:
ионная теория (теория Оствальда)
хромофорная теория
ионно-хромофорная
теория резонанса
5.3
Окраска индикаторов
Все цветные индикаторы, имеющие практическое значение,
являются слабыми органическими соединениями. Они ведут себя как слабые кислоты
или как слабые основания (индикаторы-основания способны присоединять протон, а
индикаторы-кислоты способны отщеплять протон), цвет которых в
недиссоциированном состоянии отличается от цвета их ионов. Индикатор,
рассматриваемый как кислота, будет в водном растворе находиться в равновесии со
своими ионами по уравнению:
HInd ↔Н++Ind — (1)
Здесь HInd представляет собой кислотную форму, которая
имеет «кислотную окраску», a Ind — щелочную форму, обладающую «щелочной
окраской». Применяя закон действующих масс, имеем
[H+] [Ind—] / [HInd] =KHind (2)
или
[Ind—] / [HInd] = KHind/ [H+] (3)
цвет индикатора в растворе зависит от соотyощення [Ind—] / [HInd]. Если концентрация
ионов водорода будет равна KHind, то величина [Ind—] будет равна [HInd], т. е концентрации
кислой и основной форм индикатора будут равны. В этом случае говорят, что
превращение индикатора произошло наполовину. Из этого уравнения также следует,
что наблюдаемая при изменении [Н+] перемена света индикатора
происходит не резко, а постепенно. Для каждого значения [Н+] имеется
определённое отношение концентраций обеих форм индикатора в растворе. Поскольку
имеется определённое минимальное количество каждой из форм, которое глаз может
уловить в присутствии большого количества другой формы, мы наблюдаем изменение
цвета индикатора лишь в определённых границах концентраций ионов водорода Эти
границы не имеют теоретического значения: они лишь указывают, между какими
значениями [Н+] или рН на практике наблюдается изменение цвета
индикатора. Область между двумя крайними значениями рН, определяющими эти
границы называется интервалам превращения, областью перемены окраски
индикатора, интервалом перехода, интересном изменения окраски индикаторов.
5.4
Интервал перехода индикаторов
В точке перехода можно наблюдать изменение окраски
индикатора. Но в ней не происходит резкого изменения окраски индикатора, а
изменение идёт постепенно в некоторой области значений рН. Окраска зависит от
соотношения концентраций кислотной и основной форм, что определяется
концентрацией ионов водорода в растворе. При любой концентрации ионов
водорода присутствует все формы индикатора. От ряда условий зависит
минимальная концентрация каждой из двух форм, при которой можно наблюдать одну
окраску в присутствии другой. Здесь имеет значение интенсивность обеих окрасок,
освещение, зрительная способность аналитика и т.д. Вообще говоря, два цвета
можно различить при их совместном присутствии, если концентрация одной формы
индикатора составляет 10% концентрации другой формы. Следовательно, как видно
из уравнения (1,) в случае индикаторов-кислот можно обнаружить кислотную форму
в смеси с основной, если
[H+] =Ka [HInd] / [Ind—]
= Ka1/10 (4)
и наоборот, основную форму можно наблюдать в присутствии
кислотной формы, когда [H+] =Ka10
таким образом, интервал перехода индикаторов-кислот будет при
pH=pKInd±1 (5)
Естественно, что то же выражение относится к
индикатором-основаниям IndOH — ↔ОН — Ind+
[Ind+] / [IndOH] = KIndOH/Kw
[H+] =K’ [H+] (6)
Как можно заметить из выражения (5) интервал перехода
индикаторов составляет фактически две единицы рН. В этом интервале индикатор
имеет смешанную окраску кислотной и основной форм индикатора, которая
постепенно изменяется. Для индикаторов с контрастными окрасками интервал
перехода может быть даже меньше двух единиц рН. Интервал перехода не будет
симметричных относительно значений рН если одна из форм индикатора
воспринимается легче другой. Это очень часто наблюдается в случае двухцветных
индикаторов. В действительности на интервал перехода индикатора оказывают
влияния различные факторы, поэтому в таблицах принято давать не значения показателя
степени индикатора, а практически установленные и более характерные значения
интервалов перехода.
Таблица 2. Значенне рКInd некоторых
кислотно-основных индикаторов при µ=0
|
Индикатор |
рКInd |
|
Метиловый |
3.46-0.014 |
|
Тимоловы синий |
1.65 |
|
Метиловый |
5.00-0.006 |
|
n-Нитрофенол |
7.15-0.011 |
|
Бромтиловый |
7.30 |
|
Крезоловый |
8.46 |
|
Тимоловый синий |
9.20 |
5.5 Исследование индикаторов
Пригодность индикаторов можно оценить путём установления
интервала перехода и интенсивности окраски. Для этой цели получают 0.1% или
0,01% -ный раствор индикатора в соответствующем растворителе или, если надо
получить натриевую соль в растворе гидроксида натрия. Раствор индикатора должен
быть прозрачным и содержать минимум нерастворимых примесей.
а) определение интервала перехода проводят по
следующему способу [индикаторы.] Готовят серию буферных растворов
соответственно ожидаемому интервалу перехода в десяти пробирках одинакового
цвета и диаметра, при этом значение рН в первой пробирке должно быть на 0.4 ед.
рН ниже нижнего предела, а в последней — на 0.4 единицы рН выше верхнего
предела ожидаемого интервала перехода.
б) окраски индикатора измеряют спектрофотометрическим
путём определения удельной экстинкции в максимуме кривой поглощения. Кривые
поглощения снимают при нижнем и верхнем пределах значений рН, чтобы избежать
измерения смешанных цветов:
Elcm1%=A/cd,
где А — наблюдаемая экстинкция, с — концентрация (г/100 мл), d — толщина стоя кюветы.
Результаты, полученные для новых индикаторов, следует сопоставить с данными для
чистых препаратов.
Требования, предъявляемые к кислотно-основным индикаторам:
1.
Окраска
индикатора должна быть интенсивной и различаться в кислой и щелочной среде.
2.
Изменение
окраски индикатора должно быть быстрым, чётким и обратимым.
3.
Окраска
индикатора должна изменяться в узком интервале изменения рН раствора.
4.
Индикатор
должен быть чувствительным и менять свою окраску в присутствии минимального
избытка кислоты или щелочи
5.
Индикатор
должен быть стабильным, не разлагаться з водном растворе и на воздухе.
5.6
Выбор индикатора
Выбор того или иного индикатора для титрования следует
производить на основе кривой титрования определяемого вещества. В принципе,
следует брать такой индикатор, у которого интервал превращения включает то
значение рН, которое должен иметь титруемый раствор в точке эквивалентности.
Если скачок титрования очень большой, то можно воспользоваться многими
индикаторами. Возьмем, например титрование сильной кислоты сильным основанием:
рН в точке эквивалентности равен 7, следовательно, прежде всего мы можем взять
любой индикатор, изменяющий свой цвет вблизи этого значения рН. Но если мы
рассмотрим кривую титрования 0.01н. раствор сильной кислоты, то увидим, сто рН
меняется от 5 на расстоянии 0.1% от точки эквивалентности до 9 на расстоянии
0.1% за этой точкой. Следовательно, любой индикатор, имеющий интервал перехода
в пределах рН от 5 до 9 даст результат с ошибкой 0.1%. если титруется 0.1 н
раствор кислоты, то соответствующие границы рН расширяются от 4 до 10.
Однако совсем другая ситуация складывается при титровании
слабых кислот и оснований. В этом случае в точке эквивалентности рН не равен 7
а следовательно, кривые титрования не будут симметричны относительно линии
нейтральности в общем случае можно сказать, что слабые кислоты следует
титровать в присутствии индикаторов изменяющих свой цвет в слабощелочных
растворах, а слабые основании в присутствии индикаторов, изменяющих свой цвет в
слабокислых растворах.
Для получения точных и воспроизводимых результатов анализа
необходимо соблюдать определенные условия при титровании.
. Следует устанавливать титр стандартного раствора и
применять установленный для титрования раствор в присутствии одного и того же
индикатора
. Для титрования следует брать всегда одно и то же количество
индикатора и повторять титрование определяемого вещества несколько раз до тех
пор, пока не будут подучены три близко сходящиеся результата
. Как правило, берут 1-2 капли индикатора, не забывая о том,
что индикаторы, применяемые в методе нейтрализации, сами являются кислотами и
основаниями. На нейтрализацию их также расходуется некоторое количество
раствора. Кривая титрования в присутствии очень большого количества индикатора
может иметь следующий вид. (см рис 1)
Рис 1. Кривые титрования при добавлении
а) аномально большого количества индикатора
б) необходимого количества индикатора
. Всегда следует титровать до появления одного и того же оттенка
окраски раствора, используя для титрования по возможности одинаковые объёмы
титруемого раствора.
. Необходимо выбирать такой индикатор, который изменяет свой цвет
вблизи точки эквивалентности.
. Влияние экспериментальных условий на изменение окраски
индикаторов.
. На изменение окраски органических красителей, используемых в
качестве индикаторов, оказывают влияние следующие экспериментальные условия
концентрация раствора, растворённый диоксид углерода, присутствие посторонних
солей и растворителей, белковые вещества и другие коллоиды и температура.
5.7
Влияние растворителей
Различные растворители по-разному влияют на красители,
применяемые в качестве индикаторов: в зависимости от растворителя меняется
окраска и показатель степени индикатора. В водно-этанольных растворах изменения
не так значительны, в безводных спиртовых растворах они гораздо больше. Другие
растворители могут вызывать иные явления.
В аналитической практике часто водные растворы содержат
спирт. Спирт нарушает равновесие индикаторной системы, наблюдаемый эффект
зависит не только от индикатора, но и от кислотно-основной системы раствора.
Диссоциация слабых кислот и оснований изменяется в присутствии спирта из-за
уменьшения диэлектрической константы раствора. При добавлении спирта к
сильнокислым растворам окраска индикатора-кислоты смешается в сторону кислой
окраски. Это означает, что с увеличением концентрации спирта в растворе
константа ионизации индикатора понижается: следовательно индикаторы-кислоты
становятся более чувствительными к ионом водорода (согласно формуле 4)
[H+] =Ka [HInd] / [Ind—]
Это влияние значительно меньше в растворах слабых кислот, а в
буферных растворах вообще отсутствует.
Поведение индикаторов-оснований прямо противоположное: под
влиянием добавленного спирта к сильнокислым растворам окраска
индикатора-основания смешается в сторону окраски в щелочной среде. Это
изменение ещё существеннее в слабокислых растворах и наибольшее в буферных
растворах. Например, метиловый оранжевый приобретает переходную окраску 0.01 М
водного раствора уксусной кислоты, тогда как в присутствии 40% -ного спирта
окраска ярко-жёлтая
В присутствии спирта интенсивность окраски и оттенок
индикатора также различны. Например, фенолфталеин в водных растворах гидроксида
натрия имеет зеленый цвет, а по мере прибавления спирта бледнее и приобретает
фиолетовый оттенок. Понижается и интенсивность окраски. Кроме того, в спиртовой
среде изменяется чувствительность индикаторов. Индикаторы-кислоты становятся
более чувствительными к нонам водорода, чем индикаторы-основания, независимо от
их первоначальной чувствительности
5.8
Влияние температуры
Цвет многих индикаторов зависит от температуры. Шурль нашёл,
что нагретый до кипения раствор индикатора, чувствительного к основаниям,
изменяет свою окраску в более щелочной среде, а раствор чувствительного к
кислотам индикатора — в более кислой среде. Это указывает на смещение
интервалов перевода. Так, метиловый оранжевый, который чувствителен к
основаниям, меняет свою окраску в пределах рН 3.1-4.4 при комнатной
температуре, а при 100°С интервал рН становится 2.5-3.7 это связано с тем, что
как указывалось ранее, индикаторы-основания подчиняются следующему уравнению
[Ind+] / [IndOH] = KIndOH/Kw
[H+]
Так как Кw быстрее возрастает с повышением температуры, чем
КIndOH, то для одного и того же значения [Н+] уменьшается
отношение [Ind+] / [IndOH]. Другими словами, при повышении температуры индикатор
становится более чувствительным к ионам водорода.
Следует, однако, понимать, что если индикатор сохраняет при
высокой температуре свою чувствительность к нонам водорода, то по отношению к
гидроксид ионам он будет значительно менее чувствительным, а именно в 100 раз,
т.к. Кw, при 100о в 100 раз больше Кw, при 18о, и
следовательно при 100о той же концентрации [Н+]
соответствует в 100 раз большая концентрация [OН—]. При
рассуждениях этого рода также не следует забывать, что при 100о рН
нейтрального раствора (как в рОН) равен 6.1 при высоких температурах константы
ионизации слабых кислот и основании также повышаются.
5.9
Индикаторная ошибка
Изменение цвета индикатора происходит не абсолютно в точке
эквивалентности, а раньше или позже, поэтому аналитик допускает некоторую
ошибку, прекращая титрование раньше или позже требуемого момента. Такую ошибку
называют индикаторной ошибкой, значение ее может колебаться в самых широких
пределах в зависимости от того, насколько удачно выбрал индикатор.
Если индикатор выбран правильно, то индикаторную ошибку не
принимают во внимание, если же индикатор выбран не правильно, то индикаторная
ошибка превышает допустимые погрешности и может достигать очень большой
величины.
5.10
Типы индикаторных ошибок
К индикаторным ошибкам относят такие, которые вызываются
недотитрованием или перетитрованием исследуемого раствора. В методе
нейтрализации различают несколько типов индикаторных ошибок
а) Водородная ошибка титрования, вызываемая наличием в
титруемом растворе по окончании титрования избытка ионов водорода, остающихся в
результате недотитрования сильной кислоты сильной щелочью (обозначается Н+нед
— ошибка) или перетитрования сильного основания сильной кислотой (обозначается
Н+пер-ошибка)
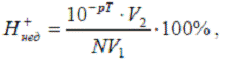
где V1 — объём в начале титрования. V2 — объём в конце
титрования.
б) Гидроксильная ошибка титрования, вызываемая наличием в
титруемом растворе по окончании титрования избытка гидроксид-ионов, остающихся
в растворе в результате недотитрования сильного основания сильной кислотой
(обозначается ОН — ошибка) или перетитрования сильной кислоты
сильной щёлочью (обозначается Н+ — ошибка)
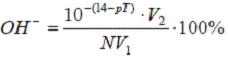
где V1 — объём в начале титрования. V2 — объём в конце
титрования
в) Кислотная ошибка титрования, вызываемая присутствием в
титруемом растворе по окончании титрования нейтральных молекул недотитрованной
слабой кислоты
![]()
г) Щелочная ошибка титрования, вызываемая присутствием в
титруемом растворе по окончанию титрования нейтральных молекул недотитрованного
слабого основания
![]()
Фенолфталеин — один из наиболее широко используемых
индикаторов, особенно при титровании слабых кислот. Он не чувствителен к
повышению температуры, ошибка от присутствия белковых веществ незначительная.
Сн (KCN) = См/ fэкв-ти = 0.1
Сн (H2SO4) = См/ fэкв—ти = 0.3/ (1/2)
= 0.6
2KCN + H2SO4 = K2SO4 + 2HCN
Пусть объем раствора KCN будет равен 10мл.
С (KCN) *V (KCN) = С (H2SO4) *V (H2SO4)(H2SO4)
= С (KCN) *V
(KCN) / С (H2SO4)
= 10*0.1/0.6 = 1.667 мл.
6.1 Расчет точек кривой титрования
1.0% добавили от необходимого объема
В растворе находится только цианид натрия, который
гидрализуется, как
KCN + H2O = KOH + HCN
CN — + H2O = OH — + HCN
Цианидный ион и HCN в растворе образуют буферную смесь. Расчет pH производим по формуле
для буферной смеси:
[OH—] =![]()
![]() =√
=√![]()
![]() ,1
,1![]()
![]() =0.000057
=0.000057
pH = 14 — pOH = 11.8337
До точки эквивалентности считаем по титруемому веществу:
2.10% добавили от необходимого объема
.667 мл — 100%
Х мл — 10%
Х=0.1667 (мл) — добавленный объем H2SO4
Получено HCN:
С (HCN) =С (H2SO4) = (Сн (H2SO4) *V (H2SO4)) / Vобщ= (0.6*0.1667) /10.1667 = = 0,009376
C (KCN) = (Сн (KCN) *V (KCN) — Сн (H2SO4) * Vдоб (H2SO4)) / Vобщ
c (KCN) = (0.051*10 — 0.6*0.1667) /10.1667
= 0.08852= pK + lg![]()
![]() = 9.3 + lg (0.08852/0,009376) = 9.0676 = 14 — pOH = 9.0676
= 9.3 + lg (0.08852/0,009376) = 9.0676 = 14 — pOH = 9.0676
.50% добавили от необходимого объема
1.667 мл — 100%
Х мл — 50%
Х = 0.8335 (мл) — добавленный объем H2SO4
Получено HCN:
С (HCN) =С (H2SO4) = (Сн (H2SO4) *V (H2SO4)) / Vобщ= (0.6*0.8335) /10.8335 = = 0.04616
C (KCN) = (Сн (KCN) *V (KCN) — Сн (H2SO4) * Vдоб (H2SO4)) / Vобщ
c (KCN) = (0.1*10 — 0.6*0.8335) /10.8335 =
0.04614= pK + lg![]()
![]() = 9.3 + lg (0.04614/0.04616) = 9.2997 = 9.2997
= 9.3 + lg (0.04614/0.04616) = 9.2997 = 9.2997
.75% добавили от необходимого объема
1.667 мл — 100%
Х мл — 75%
Х=1.25025 (мл) — добавленный объем H2SO4
Получено HCN:
С (HCN) =С (H2SO4) = (Сн (H2SO4) *V (H2SO4)) / Vобщ= (0.6*1.25025) /11.25025 = 0.06667
C (KCN) = (Сн (KCN) *V (KCN) — Сн (H2SO4) * Vдоб (H2SO4)) / Vобщ
c (KCN) = (0.1*10 — 0.6*1.25025) /11.25025
= 0.02220= pK + lg![]()
![]() = 9.3 + lg (0.011104/0.033339) = 8.8224 = 8.8224
= 9.3 + lg (0.011104/0.033339) = 8.8224 = 8.8224
.99.9% добавили от необходимого объема
1.667 мл — 100%
Х мл — 99.9%
Х=1.6653 (мл) — добавленный объем H2SO4
Получено HCN:
С (HCN) =С (H2SO4) = (Сн (H2SO4) *V (H2SO4)) / Vобщ= (0.6*1.6653) /11.6653 = 0.08565
C (KCN) = (Сн (KCN) *V (KCN) — Сн (H2SO4) * Vдоб (H2SO4)) / Vобщ
c (KCN) = (0.1*10 — 0.6*1.6653) /11.6653 =
0.00007029= pK + lg![]()
![]() = 9.3 + lg (0.0000351/0.042827) = 6.2142 = 6.2142
= 9.3 + lg (0.0000351/0.042827) = 6.2142 = 6.2142
.100% добавили от необходимого объема. Точка эквивалентности
Вещества прореагировали полностью, в растворе присутствует HCN, это слабая кислота. Рассчитываем pH:
[H+] = ![]()
![]() =
=![]()
![]() = 16.12*10-7
= 16.12*10-7
pH = 5,1245
После точки эквивалентности считаем по серной кислоте:
7.100.1% добавили от необходимого объема, раствор перетитрован
1.667 мл — 100%
Х мл — 100.1%
Х=1.668667 (мл) — добавленный объем H2SO4
[H+] = (Сн (H2SO4) * Vдоб (H2SO4) — Сн (KCN) *V (KCN)) / Vобщ
[H+] = (0.6*1.668667 — 0.1*10) /11.668667 = 0.00010285
pH = — lg [H+] = — lg0.00010285= 3.9877
.110% добавили от необходимого объема, раствор перетитрован
1.667 мл — 100%
Х мл — 100.1%
Х=1.8337 (мл) — добавленный объем H2SO4
[H+] = (Сн (H2SO4) * Vдоб (H2SO4) — Сн (KCN) *V (KCN)) / Vобщ
[H+] = (0.6*1.8337 — 0.1*10) /11.8337 = 0.008469
pH = — lg [H+] = — lg0.008469 = 2.07216
.200% добавили от необходимого объема, раствор перетитрован
1.667 мл — 100%
Х мл — 200%
Х=3.334 (мл) — добавленный объем H2SO4
[H+] = (Сн (H2SO4) * Vдоб (H2SO4) — Сн (KCN) *V (KCN)) / Vобщ
[H+] = (0.6*3.334 — 0.1*10) /13.334 = 0.07502
6.2
Построение кривой титрования
Таблица 2. Точки для построения кривой титрования по методу
«нейтрализации»
|
Процент титрования |
pH |
|
0% добавили от |
11,8337 |
|
10% добавили от |
9,0676 |
|
50% добавили от |
9,2997 |
|
75% добавили от |
8,8224 |
|
99.9% добавили |
6,2142 |
|
100% добавили |
7,1204 |
|
100.1% добавили |
3,9877 |
|
110% добавили |
2,0721 |
|
200% добавили |
1,1248 |
График 1. Кривая титрования 0,1 M KCN — 0,3 M H2SO4
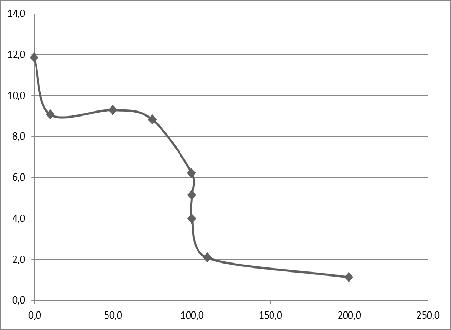
6.3
Подборка индикатора. Расчет индикаторных ошибок титрования
Таблица 4. Типы индикаторных погрешностей
|
Тип погрешности |
Причина |
Расчетная |
|
Водородная |
Избыток ионов Н+ |
∆H+ |
|
Гидроксильная |
Избыток ионов |
∆ОН |
|
Кислотная |
Избыток молекул |
∆НА = (10pK-pT*100) / (1+10pK-pT) |
|
Щелочная |
Избыток молекул |
∆МОН = = 10pK+pT-14*100 |
Тип погрешности. Причина погрешности. Расчетная формула
погрешности, %. Водородная. Избыток ионов Н+ вследствие недотитрования сильной
кислоты или перетитрования основания (сильного или слабого) сильной кислотой
∆H+ = (10-pT*V2*100) / (c*V1)
Гидроксильная. Избыток ионов ОН — вследствие недотитрования
сильного основания или перетитрования кислоты (слабой или сильной) сильным
основанием
∆ОН — = (10- (14-pT) *V2*100) / (c*V1)
Кислотная. Избыток молекул слабой кислоты НА при её
недотитровании
∆НА = (10pK-pT*100) / (1+10pK-pT)
Щелочная
Избыток молекул слабого основания MOH при его недотитровании
∆МОН = 10pK+pT-14*100
где V1 и V2 — объемы анализируемого раствора до и после титрования; с —
молярная концентрация эквивалента вещества анализируемого раствора; рК —
показатель константы диссоциации слабого электролита; рТ — показатель
титрования индикатора.
. Пусть pT розоловой кислоты = 7.1 больше pH в точке эквивалентности,
равного 7. Следовательно, раствор KCN недотитрован. Рассчитываем щелочную ошибку II-ого рода, вместо pK основания, берется pK сопряженной кислоты — pK (HCN) = 9,3: сопряженное pK = 14-9.3 = 4.7:
∆MОН = 10pK+pT-14*100
∆MОН =109,3+7,1-14*100 = 2.917*10-4 %
. Пусть pT бромтимолового синего = 6.8 меньше pH в точке
эквива-лентности, равного 7. Следовательно, раствор KCN перетитрован H2SO4. Рассчитываем водородную
ошибку (кислотную ошибку I-ого рода):
∆Н = (10—pT*V2*100) / (c*V1)
∆Н = (10-6.8* (10+1.667) *100) / (0.05*10) =
3.686*10-4 %
7.
Вывод
Роль титриметрического метода анализа не уменьшается и в наше
время. ОН является одним из самых простых. И удобных методов количественного
определения веществ. Основой данного метода является нахождение точки
эквивалентности анализируемого вещества и реагента. Ее можно определить как
физическими методами, так и с помощью индикаторов. Известно более десятка
кислотно-основных индикаторов с различными интервалами перехода, что позволяет
подобрать наиболее подходящий, для конкретной реакции. Постоянно изучаются
строение и свойства индикаторов, расширяются области их применения.
8.
Список литературы
1. Васильев
В.П. «Аналитическая химия: в двух книгах: Кн.1: Титриметрические и
гравиметрические методы анализа: Учеб. Для студ. ВУЗов, обучающихся по
химико-технологических спец. — 3-е издание, стереотип. — Москва: Дрофа, 2003год
— 368с.
2. Гильманшина
С.И. «Основы аналитической химии: курс лекций.» Доп. УМО по
направлениям пед образования РФ в качестве учебного пособия для ВУЗов С.И. —
изд. СПб, Питер 2006год-224с. (Учебное пособие)
. Крешков
А.П. «Основы аналитической химии. Теоретические основы. Количественный
анализ. «том №1. Издательство №3 (перераб.). Учебное пособие для студентов
химико-технологических спец. ВУЗов. Москва, химия, 1970год.471с.
. Лурье
Ю.Ю. «Справочник по аналитической химии.5-е издание, переработ. и доп. —
Москва: химия, 1979год-480с
. Харитонов
Ю.Я. «Аналитическая химия.». кн.1. Общие теоретические основы.
Количественный анализ: Учеб. — Москва: Высшая школа. 2001год — 615с.
. Пилипенко
A. T. Пятницкий И.В. Аналитическая химия. т.2 М: Химия. 1990-480
с.
. Харитонов
Ю. А Аналитическая химия. Аналитика, т.2 М: Высшая школа. 2001 — 560 с.
. Дорохова
Е.Н. Прохорова Г.З. Задачи и вопросы по аналитической химии М» 1984 — 216
с.